Rebetol 200mg Ribavirin Användning, biverkningar och dosering. Pris i onlineapotek. Generiska läkemedel utan recept.
Vad är Rebetol 200mg och hur används det?
Rebetol är ett receptbelagt läkemedel som används för att behandla symptomen på kronisk hepatit C. Rebetol 200 mg kan användas ensamt eller tillsammans med andra läkemedel.
Rebetol tillhör en klass av läkemedel som kallas hepatit B/hepatit C-medel; RSV-ombud.
Det är inte känt om Rebetol 200mg är säkert och effektivt för barn yngre än 3 år.
Vilka är de möjliga biverkningarna av Rebetol?
Rebetol kan orsaka allvarliga biverkningar inklusive:
- nässelfeber,
- svårt att andas,
- svullnad av ansikte, läppar, tunga eller svalg,
- synproblem
- svår smärta i övre delen av magen sprider sig till ryggen,
- illamående,
- kräkningar,
- diarre,
- ny eller förvärrad hosta,
- feber,
- stickande smärta i bröstet,
- väsande andning,
- andnöd,
- allvarlig depression,
- tankar på självskada,
- tankar på att skada andra,
- blek eller gulnad hud,
- mörkfärgad urin,
- förvirring,
- svaghet,
- frossa,
- influensaliknande symtom,
- svullna tandkött,
- munsår,
- hudsår,
- lätt att få blåmärken,
- ovanlig blödning, och
- yrsel
- Få medicinsk hjälp omedelbart om du har något av symtomen som anges ovan.
- De vanligaste biverkningarna av Rebetol inkluderar:
- illamående,
- kräkningar,
- aptitlöshet,
- feber,
- frossa,
- skakning,
- lågt antal blodkroppar,
- anemi,
- svaghet,
- trötthet,
- huvudvärk,
- träningsvärk,
- humörförändringar,
- ångest och
- irritabilitet
Tala om för läkaren om du har någon biverkning som stör dig eller som inte försvinner.
Dessa är inte alla möjliga biverkningar av Rebetol. För mer information, fråga din läkare eller apotekspersonal.
Ring din läkare för medicinsk rådgivning om biverkningar. Du kan rapportera biverkningar till FDA på 1-800-FDA-1088.
VARNING
RISK FÖR ALLVARLIGA STÖDNINGAR OCH RIBAVIRIN-ASSOCIERADE EFFEKTER
- REBETOL monoterapi är inte effektivt för behandling av kronisk hepatit C-virusinfektion och bör inte användas ensamt för denna indikation [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER].
- Den primära toxiciteten för ribavirin är hemolytisk anemi. Anemin i samband med behandling med REBETOL 200 mg kan resultera i försämring av hjärtsjukdom som har lett till dödliga och icke-dödliga hjärtinfarkter. Patienter med en anamnes på signifikant eller instabil hjärtsjukdom bör inte behandlas med REBETOL [se DOSERING OCH ADMINISTRERING, VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER samt BIVERKNINGAR].
- Signifikanta teratogena och embryocidala effekter har visats hos alla djurarter som exponerats för ribavirin. Dessutom har ribavirin en halveringstid för flera doser på 12 dagar, och det kan därför kvarstå i icke-plasmakompartment så länge som 6 månader. Därför är behandling med REBETOL kontraindicerad hos kvinnor som är gravida och hos manliga partners till kvinnor som är gravida. Extrem försiktighet måste iakttas för att undvika graviditet under behandlingen och i 6 månader efter avslutad behandling hos både kvinnliga patienter och hos kvinnliga partner till manliga patienter som tar REBETOL-behandling. Minst två tillförlitliga former av effektiva preventivmedel måste användas under behandlingen och under den 6-månaders uppföljningsperioden efter behandling [se KONTRAINDIKATIONER, VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER, Användning i specifika populationer, icke-klinisk toxikologi och PATIENTINFORMATION].
BESKRIVNING
REBETOL (ribavirin), är en syntetisk nukleosidanalog (purinanalog). Det kemiska namnet på ribavirin är 1-β-D-ribofuranosyl-1H-1,2,4-triazol-3-karboxamid och har följande strukturformel (se figur 1).
Figur 1: Strukturformel
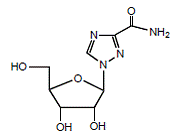
Ribavirin är ett vitt, kristallint pulver. Det är fritt lösligt i vatten och lätt lösligt i vattenfri alkohol. Den empiriska formeln är C8H12N4O5 och molekylvikten är 244,21.
REBETOL kapslar består av ett vitt pulver i en vit, ogenomskinlig gelatinkapsel. Varje kapsel innehåller 200 mg ribavirin och de inaktiva ingredienserna mikrokristallin cellulosa, laktosmonohydrat, kroskarmellosnatrium och magnesiumstearat. Kapselskalet består av gelatin, natriumlaurylsulfat, kiseldioxid och titandioxid. Kapseln är tryckt med ätbart blått farmaceutiskt bläck som är tillverkat av schellack, vattenfri etylalkohol, isopropylalkohol, n-butylalkohol, propylenglykol, ammoniumhydroxid och FD&C Blue #2 aluminiumsjö.
REBETOL 200 mg oral lösning är en klar, färglös till blek eller ljusgul vätska med bubbelgumsmak. Varje milliliter av lösningen innehåller 40 mg ribavirin och de inaktiva ingredienserna sackaros, glycerin, sorbitol, propylenglykol, natriumcitrat, citronsyra, natriumbensoat, naturlig och artificiell smak för bubbelgum #15864 och vatten.
INDIKATIONER
Kronisk hepatit C (CHC)
REBETOL® (ribavirin) i kombination med interferon alfa-2b (pegylerat och icke-pegylerat) är indicerat för behandling av kronisk hepatit C (CHC) hos patienter 3 år och äldre med kompenserad leversjukdom [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , och Användning i specifika populationer ].
Följande punkter bör beaktas när REBETOL kombinationsbehandling med PegIntron® eller INTRON A® påbörjas:
- Dessa indikationer är baserade på att uppnå odetekterbart HCV-RNA efter behandling i 24 eller 48 veckor och att bibehålla ett ihållande virologiskt svar (SVR) 24 veckor efter den sista dosen.
- Kombinationsterapi med REBETOL/PegIntron föredras framför REBETOL/INTRON A eftersom denna kombination ger avsevärt bättre svarsfrekvenser [se Kliniska studier ].
- Patienter med följande egenskaper är mindre benägna att dra nytta av ombehandling efter att en behandlingskur misslyckats: tidigare utebliven behandling, tidigare behandling med pegylerat interferon, signifikant överbryggande fibros eller cirros och genotyp 1-infektion [se Kliniska studier ].
- Inga säkerhets- och effektdata finns tillgängliga för behandling längre än ett år.
DOSERING OCH ADMINISTRERING
Under inga omständigheter får REBETOL kapslar öppnas, krossas eller krossas. REBETOL 200 mg ska tas med mat [se KLINISK FARMAKOLOGI ]. REBETOL ska inte användas till patienter med kreatininclearance mindre än 50 ml/min.
REBETOL/PegIntron kombinationsterapi
Vuxna patienter
Den rekommenderade dosen av PegIntron är 1,5 mikrogram/kg/vecka subkutant i kombination med 800 till 1400 mg REBETOL 200 mg kapslar oralt baserat på patientens kroppsvikt (se tabell 1). Volymen av PegIntron som ska injiceras beror på styrkan av PegIntron och patientens kroppsvikt, se märkningen för PegIntron för ytterligare doseringsinformation.
Behandlingslängd – Interferon alfa-naiva patienter
Behandlingstiden för patienter med genotyp 1 är 48 veckor. Avbrytande av behandlingen bör övervägas hos patienter som inte uppnår minst 2 log10 fall eller förlust av HCV-RNA efter 12 veckor, eller om HCV-RNA fortfarande kan detekteras efter 24 veckors behandling. Patienter med genotyp 2 och 3 ska behandlas i 24 veckor.
Behandlingens varaktighet – Ombehandling med PegIntron/REBETOL av tidigare behandlingsmisslyckanden
Behandlingslängden för patienter som tidigare misslyckats med behandlingen är 48 veckor, oavsett HCV-genotyp. Återbehandlade patienter som inte lyckas uppnå odetekterbart HCV-RNA vid vecka 12 av behandlingen, eller vars HCV-RNA förblir detekterbart efter 24 veckors behandling, är högst osannolikt att uppnå SVR och avbrytande av behandlingen bör övervägas [se Kliniska studier ].
Pediatriska patienter
Dosering för pediatriska patienter bestäms av kroppsyta för PegIntron och av kroppsvikt för REBETOL. Den rekommenderade dosen av PegIntron är 60 mcg/m²/vecka subkutant i kombination med 15 mg/kg/dag av REBETOL 200 mg oralt i två uppdelade doser (se tabell 2) för pediatriska patienter i åldrarna 3-17 år. Patienter som fyller 18 år samtidigt som de får PegIntron/REBETOL 200 mg bör fortsätta med den pediatriska doseringsregimen. Behandlingstiden för patienter med genotyp 1 är 48 veckor. Patienter med genotyp 2 och 3 ska behandlas i 24 veckor.
REBETOL/INTRON En kombinationsterapi
Vuxna
Behandlingslängd – Interferon alfa-naiva patienter
Den rekommenderade dosen av INTRON A är 3 miljoner IE tre gånger i veckan subkutant. Den rekommenderade dosen av REBETOL kapslar beror på patientens kroppsvikt (se tabell 3). Den rekommenderade behandlingstiden för patienter som tidigare inte behandlats med interferon är 24 till 48 veckor. Behandlingslängden bör anpassas till patienten beroende på sjukdomens baslinjeegenskaper, behandlingssvar och behandlingens tolerabilitet [se INDIKATIONER OCH ANVÄNDNING , NEGATIVA REAKTIONER , och Kliniska studier ]. Efter 24 veckors behandling bör det virologiska svaret utvärderas. Avbrytande av behandlingen bör övervägas hos alla patienter som inte har uppnått ett HCV-RNA under analysens detektionsgräns inom 24 veckor. Det finns inga säkerhets- och effektdata för behandling under längre tid än 48 veckor i den tidigare obehandlade patientpopulationen.
Behandlingens varaktighet – Ombehandling med INTRON A/REBETOL hos återfallspatienter
Hos patienter som återfaller efter monoterapi med icke-pegylerat interferon är den rekommenderade behandlingstiden 24 veckor.
Pediatrik
Den rekommenderade dosen av REBETOL är 15 mg/kg per dag oralt (delad dos AM och PM). Se tabell 2 för pediatrisk dosering av REBETOL i kombination med INTRON A. INTRON A för injektion efter kroppsvikt på 25 kg till 61 kg är 3 miljoner IE/m² tre gånger i veckan subkutant. Se doseringstabell för vuxna för mer än 61 kg kroppsvikt.
Rekommenderad behandlingslängd är 48 veckor för pediatriska patienter med genotyp 1. Efter 24 veckors behandling bör det virologiska svaret utvärderas. Avbrytande av behandlingen bör övervägas hos alla patienter som inte har uppnått ett HCV-RNA under analysens detektionsgräns vid denna tidpunkt. Den rekommenderade behandlingstiden för pediatriska patienter med genotyp 2/3 är 24 veckor.
Laboratorietester
Följande laboratorietester rekommenderas för alla patienter som behandlas med REBETOL 200 mg, innan behandlingen påbörjas och sedan med jämna mellanrum.
Standard hematologiska tester - inklusive hemoglobin (förbehandling, vecka 2 och vecka 4 av behandlingen, och som kliniskt lämpligt [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ], fullständigt och differentiellt antal vita blodkroppar och antal blodplättar.
- Blodkemi - leverfunktionstester och TSH.
- Graviditet - inklusive månatlig övervakning för fertila kvinnor.
- EKG [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Dosändringar
Om allvarliga biverkningar eller laboratorieavvikelser utvecklas under kombinationsbehandling med REBETOL/INTRON A eller REBETOL/PegIntron, modifiera eller avbryt dosen tills biverkningen avtar eller minskar i svårighetsgrad [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. Om intoleransen kvarstår efter dosjustering ska kombinationsbehandlingen avbrytas. Dosreduktion av PegIntron hos vuxna patienter på REBETOL/PegIntron kombinationsbehandling åstadkommes i en tvåstegsprocess från den ursprungliga startdosen på 1,5 mikrogram/kg/vecka, till 1 mikrogram/kg/vecka, sedan till 0,5 mikrogram/kg/vecka. , om det behövs. Se märkningen för PegIntron för ytterligare information om dosminskning av PegIntron.
I studie 2 med kombinationsterapi för vuxna inträffade dosminskningar hos 42 % av försökspersonerna som fick PegIntron 1,5 mikrogram/kg och REBETOL 800 mg dagligen, inklusive 57 % av de försökspersoner som vägde 60 kg eller mindre. I studie 4 hade 16 % av försökspersonerna en dosreduktion av PegIntron till 1 mcg/kg i kombination med REBETOL, med ytterligare 4 % som krävde den andra dosen av PegIntron till 0,5 mcg/kg på grund av biverkningar [se NEGATIVA REAKTIONER ].
Dosreduktion hos pediatriska patienter uppnås genom att modifiera den rekommenderade dosen av PegIntron i en tvåstegsprocess från den ursprungliga startdosen på 60 mcg/m²/vecka, till 40 mcg/m²/vecka, sedan till 20 mcg/m²/vecka, om behövs (se tabell 4). I den pediatriska kombinationsterapistudien inträffade dosminskningar hos 25 % av försökspersonerna som fick PegIntron 60 mikrogram/m² per vecka och REBETOL 15 mg/kg dagligen. Dosreduktion hos pediatriska patienter uppnås genom att modifiera den rekommenderade REBETOL-dosen från den ursprungliga startdosen på 15 mg/kg dagligen i en tvåstegsprocess till 12 mg/kg/dag, sedan till 8 mg/kg/dag, vid behov ( se tabell 4).
REBETOL 200 mg ska inte användas till patienter med kreatininclearance mindre än 50 ml/min. Patienter med nedsatt njurfunktion och de över 50 år bör övervakas noggrant med avseende på utveckling av anemi [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , Användning i specifika populationer , och KLINISK FARMAKOLOGI ].
REBETOL 200 mg ska administreras med försiktighet till patienter med redan existerande hjärtsjukdom. Patienterna bör utvärderas innan behandlingen påbörjas och bör övervakas på lämpligt sätt under behandlingen. Om det finns någon försämring av kardiovaskulär status ska behandlingen avbrytas [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
För patienter med en anamnes på stabil kardiovaskulär sjukdom krävs en permanent dosreduktion om hemoglobinet minskar med mer än eller lika med 2 g/dL under någon 4-veckorsperiod. Om hemoglobinet förblir mindre än 12 g/dL efter 4 veckor med en reducerad dos, bör patienten dessutom avbryta kombinationsbehandlingen för dessa hjärtpatienter.
Det rekommenderas att en patient vars hemoglobinnivå faller under 10 g/dL får sin REBETOL-dos modifierad eller utsätts enligt Tabell 4 [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Se märkningen för INTRON A eller PegIntron för ytterligare information om hur man minskar en INTRON A- eller PegIntron-dos.
Avbrytande av dosering
Vuxna
Hos HCV genotyp 1, interferon-alfa-naiva patienter som får PegIntron i kombination med ribavirin, rekommenderas att behandlingen avbryts om det inte finns minst 2 log10 droppar eller förlust av HCV-RNA efter 12 veckors behandling, eller om HCV-RNA nivåerna förblir detekterbara efter 24 veckors behandling. Oavsett genotyp är det högst osannolikt att tidigare behandlade patienter som har detekterbart HCV-RNA vid vecka 12 eller 24 uppnår SVR och avbrytande av behandlingen bör övervägas.
Pediatrik (3-17 år)
Det rekommenderas att patienter som får PegIntron/REBETOL-kombinationen (exklusive HCV genotyp 2 och 3) avbryts från behandlingen vid 12 veckor om deras behandling Vecka 12 HCV-RNA sjunkit mindre än 2 log10 jämfört med en förbehandling eller vid 24 veckor om de har detekterbar HCV-RNA vid behandling vecka 24.
HUR LEVERERAS
Doseringsformer och styrkor
REBETOL 200mg Kapslar 200 mg
REBETOL 200 mg oral lösning 40 mg per ml
Förvaring Och Hantering
REBETOL 200 mg kapslar är vita, ogenomskinliga kapslar med REBETOL, 200 mg, och Schering Corporation-logotypen präglad på kapselhöljet; kapslarna är förpackade i en flaska som innehåller 56 kapslar ( NDC 0085-1351-05), 70 kapslar ( NDC 0085-1385-07), och 84 kapslar ( NDC 0085-1194-03).
REBETOL 200mg oral lösning 40 mg per ml är en klar, färglös till blek eller ljusgul vätska med bubbelgumsmak och den är förpackad i 4-oz bärnstensfärgade glasflaskor (100 ml/flaska) med barnsäkra förslutningar ( NDC 0085-1318-01).
Flaskan med REBETOL kapslar ska förvaras vid 25°C (77°F); utflykter tillåtna till 15-30°C (59-86°F) [se USP-kontrollerad rumstemperatur ].
REBETOL 200 mg oral lösning ska förvaras mellan 2-8°C (36-46°F) eller vid 25°C (77°F); utflykter tillåtna till 15-30°C (59-86°F) [se USP-kontrollerad rumstemperatur ].
REBETOL 200mg oral lösning tillverkad för: Merck Sharp & Dohme Corp., ett dotterbolag till Whitehouse Station, NJ 08889, USA. Tillverkade av: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Kanada REBETOL Kapslar tillverkade av: Merck Sharp & Dohme Corp., ett dotterbolag till Whitehouse Station, NJ 08889, USA. Reviderad: dec 2014.
BIEFFEKTER
Kliniska prövningar med REBETOL i kombination med PegIntron eller INTRON A har utförts på över 7800 försökspersoner från 3 till 76 år.
Den primära toxiciteten för ribavirin är hemolytisk anemi. Minskningar av hemoglobinnivåer inträffade inom de första 1 till 2 veckorna av oral behandling. Hjärt- och lungreaktioner associerade med anemi förekom hos cirka 10 % av patienterna [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Mer än 96 % av alla försökspersoner i kliniska prövningar upplevde en eller flera biverkningar. De vanligaste rapporterade biverkningarna hos vuxna försökspersoner som fick PegIntron eller INTRON A i kombination med REBETOL 200 mg var inflammation/reaktion på injektionsstället, trötthet/asteni, huvudvärk, stelhet, feber, illamående, myalgi och ångest/emotionell labilitet/irritabilitet. De vanligaste biverkningarna hos pediatriska försökspersoner i åldrarna 3 och äldre som fick REBETOL 200 mg i kombination med PegIntron eller INTRON A var pyrexi, huvudvärk, neutropeni, trötthet, anorexi, erytem på injektionsstället och kräkningar.
Avsnittet Biverkningar hänvisar till följande kliniska prövningar:
- REBETOL/PegIntron Kombinationsterapiförsök:
- Klinisk studie 1 – utvärderad monoterapi med PegIntron (beskrivs inte ytterligare i denna etikett; se märkning för PegIntron för information om denna studie).
- Studie 2 – utvärderad REBETOL 800 mg/dag platt dos i kombination med 1,5 mikrogram/kg/vecka PegIntron eller med INTRON A.
- Studie 3 – utvärderade PegIntron/viktbaserad REBETOL 200mg i kombination med PegIntron/plattdos REBETOL 200mg regim.
- Studie 4 – jämförde två doser av PegIntron (1,5 mcg/kg/vecka och 1 mcg/kg/vecka) i kombination med REBETOL och en tredje behandlingsgrupp som fick Pegasys® (180 mcg/vecka)/Copegus® (1000-1200 mg/dag) ).
- Studie 5 – utvärderade PegIntron (1,5 mikrogram/kg/vecka) i kombination med viktbaserad REBETOL hos patienter med tidigare behandlingssvikt.
- PegIntron/REBETOL 200 mg kombinationsterapi hos pediatriska patienter
- REBETOL/INTRON A kombinationsterapiförsök för vuxna och pediatrik
Allvarliga biverkningar har inträffat hos cirka 12 % av försökspersonerna i kliniska prövningar med PegIntron med eller utan REBETOL [se LÅDA VARNING , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. De vanligaste allvarliga händelserna som inträffade hos patienter som behandlades med PegIntron och REBETOL 200 mg var depression och självmordstankar [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ], var och en förekommer med en frekvens på mindre än 1 %. Självmordstankar eller självmordsförsök förekom oftare bland pediatriska patienter, främst ungdomar, jämfört med vuxna patienter (2,4 % mot 1 %) under behandling och uppföljning utanför behandlingen [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. Den vanligaste dödliga reaktionen som inträffade hos patienter som behandlades med PegIntron och REBETOL 200 mg var hjärtstillestånd, självmordstankar och självmordsförsök [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ], alla förekommer hos mindre än 1 % av försökspersonerna.
Eftersom kliniska prövningar genomförs under vitt skilda förhållanden, kan biverkningsfrekvensen som observerats i de kliniska prövningarna av ett läkemedel inte direkt jämföras med frekvensen i de kliniska prövningarna av ett annat läkemedel och återspeglar kanske inte frekvensen som observerats i klinisk praxis.
Erfarenhet av kliniska prövningar – REBETOL/PegIntron-kombinationsterapi
Vuxna ämnen
Biverkningar som inträffade i den kliniska prövningen med mer än 5 % incidens ges av behandlingsgrupp från REBETOL/PegIntron-kombinationsterapin (studie 2) i tabell 5.
Tabell 6 sammanfattar de behandlingsrelaterade biverkningarna i studie 4 som inträffade med en incidens på mer än eller lika med 10 %.
Incidensen av allvarliga biverkningar var jämförbar i alla studier. I studie 3 rapporterades en liknande incidens av allvarliga biverkningar för den viktbaserade REBETOL-gruppen (12 %) och för REBETOL-regimen med platt dos. I studie 2 var incidensen av allvarliga biverkningar 17 % i PegIntron/REBETOL-grupperna jämfört med 14 % i INTRON A/REBETOL-gruppen.
många men inte alla fall försvann biverkningarna efter dosreduktion eller avbrytande av behandlingen. Vissa försökspersoner upplevde pågående eller nya allvarliga biverkningar under den 6 månader långa uppföljningsperioden. I studie 2 fortsatte många försökspersoner att uppleva biverkningar flera månader efter avslutad behandling. Vid slutet av den 6 månader långa uppföljningsperioden var incidensen av pågående biverkningar efter kroppsklass i gruppen PegIntron 1,5/REBETOL 200 mg 33 % (psykiatrisk), 20 % (muskuloskeletala) och 10 % (för endokrina och för GI). Hos cirka 10 till 15 % av försökspersonerna hade viktminskning, trötthet och huvudvärk inte försvunnit.
Det har förekommit 31 försökspersoners dödsfall som inträffade under behandling eller under uppföljning i dessa kliniska prövningar. I studie 1 var det 1 självmord hos en patient som fick PegIntron monoterapi och 2 dödsfall bland patienter som fick INTRON A monoterapi (1 mord/självmord och 1 plötslig död). I studie 2 var det 1 självmord hos en patient som fick PegIntron/REBETOL 200 mg kombinationsbehandling; och 1 försöksperson döds i gruppen INTRON A/REBETOL 200 mg (motorfordonsolycka). I studie 3 var det 14 dödsfall, varav 2 var troliga självmord och 1 var ett oförklarat dödsfall hos en person med en relevant medicinsk historia av depression. I studie 4 var det 12 dödsfall, varav 6 inträffade hos försökspersoner som fick PegIntron/REBETOL kombinationsterapi, 5 i PegIntron 1,5 mcg/REBETOL-armen (N=1019) och 1 i PegIntron 1 mcg/REBETOL-armen (N= 1016), och 6 av dessa inträffade hos patienter som fick Pegasys/Copegus (N=1035); det var 3 självmord som inträffade under uppföljningsperioden utanför behandlingen hos försökspersoner som fick PegIntron (1,5 mikrogram/kg)/REBETOL 200 mg kombinationsbehandling.
studier 1 och 2 avbröt 10 till 14 % av försökspersonerna som fick PegIntron, ensamt eller i kombination med REBETOL 200 mg, behandlingen jämfört med 6 % som behandlades med enbart INTRON A och 13 % som behandlades med INTRON A i kombination med REBETOL. På liknande sätt i studie 3 avbröt 15 % av försökspersonerna som fick PegIntron i kombination med viktbaserad REBETOL och 14 % av försökspersonerna som fick PegIntron och en flatdos REBETOL 200 mg behandlingen på grund av en biverkning. De vanligaste orsakerna till att behandlingen avbröts var relaterade till kända interferoneffekter av psykiatriska, systemiska (t.ex. trötthet, huvudvärk) eller gastrointestinala biverkningar. I studie 4 avbröt 13 % av försökspersonerna i PegIntron 1,5 mcg/REBETOL 200 mg-armen, 10 % i PegIntron 1 mcg/REBETOL 200 mg-armen och 13 % i Pegasys 180 mcg/Copegus-armen på grund av biverkningar.
studie 2 inträffade dosminskningar på grund av biverkningar hos 42 % av försökspersonerna som fick PegIntron (1,5 mcg/kg)/REBETOL 200 mg och hos 34 % av de som fick INTRON A/REBETOL. Majoriteten av försökspersonerna (57 %) som vägde 60 kg eller mindre som fick PegIntron (1,5 mikrogram/kg)/REBETOL krävde dosreduktion. Reduktionen av interferon var dosrelaterad (PegIntron 1,5 mcg/kg högre än PegIntron 0,5 mcg/kg eller INTRON A), 40%, 27% respektive 28%. Dosminskningen för REBETOL 200 mg var likartad i alla tre grupperna, 33 till 35 %. De vanligaste orsakerna till dosändringar var neutropeni (18 %) eller anemi (9 %) (se Laboratorievärden ). Andra vanliga orsaker var depression, trötthet, illamående och trombocytopeni. I studie 3 inträffade dosändringar på grund av biverkningar oftare med viktbaserad dosering (WBD) jämfört med platt dosering (29 % respektive 23 %). I studie 4 hade 16 % av försökspersonerna en dosreduktion av PegIntron till 1 mcg/kg i kombination med REBETOL 200 mg, med ytterligare 4 % som krävde den andra dosen av PegIntron till 0,5 mcg/kg på grund av biverkningar jämfört med 15 % av försökspersoner i Pegasys/Copegus-armen, som krävde en dosreduktion till 135 mikrogram/vecka med Pegasys, med ytterligare 7 % i Pegasys/Copegus-armen som krävde en andra dosreduktion till 90 mikrogram/vecka med Pegasys.
kombinationsstudierna med PegIntron/REBETOL 200 mg var de vanligaste biverkningarna psykiatriska, vilka inträffade bland 77 % av försökspersonerna i studie 2 och 68 % till 69 % av försökspersonerna i studie 3. Dessa psykiatriska biverkningar inkluderade oftast depression, irritabilitet och sömnlöshet, var och en rapporterad av cirka 30 % till 40 % av försökspersonerna i alla behandlingsgrupper. Suicidalt beteende (idéer, försök och självmord) förekom hos 2 % av alla försökspersoner under behandling eller under uppföljning efter avslutad behandling [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. I studie 4 inträffade psykiatriska biverkningar hos 58 % av försökspersonerna i PegIntron 1,5 mcg/REBETOL 200 mg-armen, 55 % av försökspersonerna i PegIntron 1 mcg/REBETOL 200 mg-armen och 57 % av försökspersonerna i Pegasys-armen/180 mcg. .
PegIntron inducerade trötthet eller huvudvärk hos ungefär två tredjedelar av försökspersonerna, med feber eller stelhet hos ungefär hälften av försökspersonerna. Svårighetsgraden av några av dessa systemiska symtom (t.ex. feber och huvudvärk) tenderade att minska när behandlingen fortsatte. I studierna 1 och 2 inträffade inflammation och reaktion på appliceringsstället (t.ex. blåmärken, klåda och irritation) med ungefär dubbelt så hög incidens med PegIntron-behandlingar (hos upp till 75 % av försökspersonerna) jämfört med INTRON A. Smärta på injektionsstället var dock sällan (2 till 3%) i alla grupper. I studie 3 var det en total incidens på 23 % till 24 % för reaktioner på injektionsstället eller inflammation.
Patienter som fick REBETOL/PegIntron som återbehandling efter att ha misslyckats med en tidigare interferonkombinationsregim rapporterade biverkningar liknande de som tidigare associerats med denna regim under kliniska prövningar av behandlingsnaiva försökspersoner.
Pediatriska ämnen
allmänhet var biverkningsprofilen i den pediatriska populationen liknande den som observerats hos vuxna. I den pediatriska studien var de vanligaste biverkningarna hos alla försökspersoner pyrexi (80 %), huvudvärk (62 %), neutropeni (33 %), trötthet (30 %), anorexi (29 %), erytem på injektionsstället (29 %). %) och kräkningar (27 %). Majoriteten av biverkningarna som rapporterades i studien var milda eller måttliga i svårighetsgrad. Allvarliga biverkningar rapporterades hos 7 % (8/107) av alla försökspersoner och inkluderade smärta på injektionsstället (1 %), smärta i extremiteter (1 %), huvudvärk (1 %), neutropeni (1 %) och pyrexi (4 %). %). Viktiga biverkningar som inträffade i denna patientpopulation var nervositet (7%; 7/107), aggression (3%; 3/107), ilska (2%; 2/107) och depression (1%; 1/107) . Fem försökspersoner fick levotyroxinbehandling, tre med klinisk hypotyreos och två med asymtomatiska TSH-förhöjningar. Vikt- och höjdökning hos pediatriska patienter som behandlats med PegIntron plus REBETOL släpade efter den som förutspåtts av normativa populationsdata för hela behandlingens längd. Allvarligt hämmad tillväxthastighet (mindre än 3:e percentilen) observerades hos 70 % av försökspersonerna under behandling.
Dosjusteringar av PegIntron och/eller ribavirin krävdes hos 25 % av försökspersonerna på grund av behandlingsrelaterade biverkningar, oftast för anemi, neutropeni och viktminskning. Två försökspersoner (2 %; 2/107) avbröt behandlingen till följd av en biverkning.
Biverkningar som inträffade med en incidens på mer än eller lika med 10 % hos de pediatriska försökspersonerna anges i Tabell 7.
Nittiofyra av 107 försökspersoner ingick i en 5-årig långtidsuppföljningsstudie. Långtidseffekterna på tillväxten var mindre hos de patienter som behandlades i 24 veckor än de som behandlades i 48 veckor. Tjugofyra procent av försökspersonerna (11/46) som behandlades i 24 veckor och 40% av försökspersonerna (19/48) som behandlades i 48 veckor hade en minskning med > 15 percentilen i höjd för ålder från förbehandling till slutet av 5 år långtidsuppföljning jämfört med baseline-percentiler före behandlingen. Elva procent av försökspersonerna (5/46) som behandlats i 24 veckor och 13% av försökspersonerna (6/48) som behandlats i 48 veckor observerades ha en minskning från förbehandlingens baslinje med > 30 längd-för-ålder-percentiler till slutet av den 5-åriga långtidsuppföljningen. Även om den observerades i alla åldersgrupper, verkade den högsta risken för minskad längd i slutet av långtidsuppföljningen korrelera med initiering av kombinationsbehandling under åren med förväntad maximal tillväxthastighet. [Ser VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]
Laboratorievärden
Vuxna och pediatriska ämnen
Biverkningsprofilen i studie 3, som jämförde PegIntron/viktbaserad REBETOL 200 mg kombination med en PegIntron/platt dos REBETOL 200 mg regim, avslöjade en ökad andel anemi med viktbaserad dosering (29 % mot 19 % för viktbaserad dosering). vs plandosregimer, respektive). Men majoriteten av fallen av anemi var milda och svarade på dosminskningar.
Förändringar i utvalda laboratorievärden under behandling i kombination med REBETOL 200 mg behandling beskrivs nedan. Minskning av hemoglobin, leukocyter, neutrofiler och blodplättar kan kräva dosreduktion eller permanent avbrytande av behandlingen [ser DOSERING OCH ADMINISTRERING ]. Förändringar i utvalda laboratorievärden under behandlingen beskrivs i Tabell 8. De flesta förändringarna i laboratorievärden i PegIntron/REBETOL 200 mg-studien med pediatrik var milda eller måttliga.
Hemoglobin
Hemoglobinnivåerna minskade till mindre än 11 g/dL hos cirka 30 % av försökspersonerna i studie 2. I studie 3 hade 47 % av försökspersonerna som fick WBD REBETOL 200 mg och 33 % på en platt dos REBETOL 200 mg minskningar i hemoglobinnivåer på mindre än 11 g /dl. Minskning av hemoglobin till mindre än 9 g/dL förekom oftare hos patienter som fick WBD jämfört med platt dosering (4 % respektive 2 %). I studie 2 krävdes dosändring hos 9 % och 13 % av försökspersonerna i grupperna PegIntron/REBETOL 200 mg och INTRON A/REBETOL 200 mg. I studie 4 hade försökspersoner som fick PegIntron (1,5 mcg/kg)/REBETOL minskningar av hemoglobinnivåerna till mellan 8,5 till mindre än 10 g/dL (28 %) och till mindre än 8,5 g/dL (3 %), medan det hos patienter Efter att ha fått Pegasys 180 mcg/Copegus inträffade dessa minskningar hos 26 % respektive 4 % av försökspersonerna. Hemoglobinnivåerna blev stabila vid behandlingsvecka 4-6 i genomsnitt. Det typiska mönstret som observerades var en minskning av hemoglobinnivåerna vid behandling vecka 4 följt av stabilisering och en platå, som bibehölls till slutet av behandlingen. I PegIntron monoterapistudien var hemoglobinminskningen i allmänhet mild och dosjusteringar var sällan nödvändiga [se DOSERING OCH ADMINISTRERING ].
Neutrofiler
Minskning av antalet neutrofiler observerades hos en majoritet av vuxna patienter som behandlades med kombinationsbehandling med REBETOL 200 mg i studie 2 (85 %) och INTRON A/REBETOL (60 %). Allvarlig potentiellt livshotande neutropeni (mindre än 0,5 x 109/L) förekom hos 2 % av försökspersonerna som behandlades med INTRON A/REBETOL 200 mg och hos cirka 4 % av försökspersonerna som behandlades med PegIntron/REBETOL 200 mg i studie 2. Arton procent av försökspersonerna PegIntron/REBETOL i studie 2 krävde modifiering av interferondosen. Få försökspersoner (mindre än 1%) krävde permanent avbrytande av behandlingen. Neutrofilantalet återgick i allmänhet till nivåerna före behandling 4 veckor efter avslutad behandling [se DOSERING OCH ADMINISTRERING ].
Blodplättar
Trombocytantalet minskade till mindre än 100 000/mm³ hos cirka 20 % av patienterna som behandlades med PegIntron enbart eller med REBETOL 200 mg och hos 6 % av vuxna patienter som behandlades med INTRON A/REBETOL. Allvarlig minskning av trombocytantalet (mindre än 50 000/mm³) förekommer hos mindre än 4 % av vuxna försökspersoner. Patienter kan behöva avbrytande eller dosjustering till följd av trombocytminskning [se DOSERING OCH ADMINISTRERING ]. I studie 2 krävde 1 % eller 3 % av försökspersonerna dosjustering av INTRON A respektive PegIntron. Trombocytantalet återgick i allmänhet till nivåerna före behandling 4 veckor efter avslutad behandling.
Sköldkörtelfunktion
Utveckling av TSH-avvikelser, med eller utan kliniska manifestationer, är associerad med interferonterapier. I studie 2 uppträdde kliniskt uppenbara sköldkörtelstörningar bland patienter som behandlades med antingen INTRON A eller PegIntron (med eller utan REBETOL) med liknande incidens (5 % för hypotyreos och 3 % för hypertyreos). Försökspersonerna utvecklade nya TSH-avvikelser under behandlingen och under uppföljningsperioden. Vid slutet av uppföljningsperioden hade 7 % av försökspersonerna fortfarande onormala TSH-värden.
Bilirubin och urinsyra
I studie 2 utvecklade 10 till 14 % av försökspersonerna hyperbilirubinemi och 33 till 38 % utvecklade hyperurikemi i samband med hemolys. Sex försökspersoner utvecklade mild till måttlig gikt.
Erfarenhet av kliniska prövningar – REBETOL/INTRON En kombinationsterapi
Vuxna ämnen
kliniska prövningar avbröt 19 % och 6 % av tidigare obehandlade respektive patienter med återfall behandlingen på grund av biverkningar i kombinationsarmarna jämfört med 13 % och 3 % i interferonarmarna. Utvalda behandlingsrelaterade biverkningar som inträffade i de amerikanska studierna med mer än eller lika med 5 % incidens tillhandahålls per behandlingsgrupp (se tabell 9). I allmänhet rapporterades de utvalda behandlingsrelaterade biverkningarna med lägre incidens i de internationella studierna jämfört med de amerikanska studierna, med undantag för asteni, influensaliknande symtom, nervositet och klåda.
Pediatriska ämnen
kliniska prövningar av 118 pediatriska försökspersoner i åldern 3 till 16 år avbröt 6 % behandlingen på grund av biverkningar. Dosjusteringar krävdes hos 30 % av försökspersonerna, vanligast för anemi och neutropeni. I allmänhet var biverkningsprofilen i den pediatriska populationen liknande den som observerats hos vuxna. Störningar på injektionsstället, feber, anorexi, kräkningar och känslomässig labilitet förekom oftare hos pediatriska försökspersoner jämfört med vuxna. Omvänt upplevde pediatriska försökspersoner mindre trötthet, dyspepsi, artralgi, sömnlöshet, irritabilitet, försämrad koncentration, dyspné och klåda jämfört med vuxna försökspersoner. Utvalda behandlingsrelaterade biverkningar som inträffade med mer än eller lika med 5 % incidens bland alla pediatriska försökspersoner som fick den rekommenderade dosen av REBETOL/INTRON A-kombinationsbehandling ges i Tabell 9.
Under en 48-veckors behandlingskur skedde en minskning av hastigheten för linjär tillväxt (genomsnittlig procentiltilldelningsminskning med 7 %) och en minskning av hastigheten för viktökning (genomsnittlig percentiltilldelningsminskning med 9 %). En allmän vändning av dessa trender noterades under den 24 veckor långa perioden efter behandling. Långtidsdata från ett begränsat antal patienter tyder dock på att kombinationsbehandling kan inducera en tillväxthämning som resulterar i minskad slutlig vuxenlängd hos vissa patienter [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Laboratorievärden
Förändringar i utvalda hematologiska värden (hemoglobin, vita blodkroppar, neutrofiler och blodplättar) under behandlingen beskrivs nedan (se tabell 10).
Hemoglobin. Hemoglobinminskningar bland försökspersoner som fick REBETOL-behandling började vid vecka 1, med stabilisering vid vecka 4. Hos tidigare obehandlade försökspersoner som behandlats i 48 veckor var den genomsnittliga maximala minskningen från baslinjen 3,1 g/dL i den amerikanska studien och 2,9 g/dL i den internationella rättegång. Hos patienter med återfall var den genomsnittliga maximala minskningen från baslinjen 2,8 g/dL i den amerikanska studien och 2,6 g/dL i den internationella studien. Hemoglobinvärdena återgick till nivåerna före behandling inom 4 till 8 veckor efter avslutad behandling hos de flesta patienter.
Bilirubin och urinsyra. Ökning av både bilirubin och urinsyra, associerad med hemolys, noterades i kliniska prövningar. De flesta var måttliga biokemiska förändringar och reverserades inom 4 veckor efter avslutad behandling. Denna observation inträffade oftast hos personer med en tidigare diagnos av Gilberts syndrom. Detta har inte associerats med leverdysfunktion eller klinisk morbiditet.
Upplevelser efter marknadsföring
Följande biverkningar har identifierats och rapporterats under användning av REBETOL i kombination med INTRON A eller PegIntron efter godkännande. Eftersom dessa reaktioner rapporteras frivilligt från en population av osäker storlek är det inte alltid möjligt att på ett tillförlitligt sätt uppskatta deras frekvens eller fastställa ett orsakssamband till läkemedelsexponering.
Blod- och lymfsystemet
Aplasi av ren röda blodkroppar, aplastisk anemi
Öron- och labyrintsjukdomar
Hörselstörning, svindel
Andningsvägar, bröstkorg och mediastinum
Pulmonell hypertoni
Ögonstörningar
Serös näthinneavlossning
Endokrina störningar
Diabetes
LÄKEMEDELSINTERAKTIONER
Didanosin
Exponering för didanosin eller dess aktiva metabolit (dideoxiadenosin 5'-trifosfat) ökar när didanosin administreras samtidigt med ribavirin, vilket kan orsaka eller förvärra klinisk toxicitet; Därför är samtidig administrering av REBETOL 200 mg kapslar eller oral lösning och didanosin kontraindicerat. Rapporter om fatal leversvikt, såväl som perifer neuropati, pankreatit och symptomatisk hyperlaktatemi/laktacidos har rapporterats i kliniska prövningar.
Nukleosidanaloger
Leverdekompensation (viss dödlig) har inträffat hos cirrhotiska HIV/HCV-infekterade patienter som fått antiretroviral kombinationsbehandling för HIV och interferon alfa och ribavirin. Tillägg av behandling med enbart alfa-interferoner eller i kombination med ribavirin kan öka risken i denna patientpopulation. Patienter som får interferon med ribavirin och nukleosid omvänt transkriptashämmare (NRTI) bör övervakas noggrant med avseende på behandlingsrelaterade toxiciteter, särskilt leverdekompensation och anemi. Avbrytande av NRTI bör anses vara medicinskt lämpligt (se märkning för individuell NRTI-produkt ). Dosreduktion eller utsättande av interferon, ribavirin eller båda bör också övervägas om förvärrade kliniska toxiciteter observeras, inklusive leverdekompensation (t.ex. Child-Pugh större än 6).
Ribavirin kan motverka cellkulturens antivirala aktivitet av stavudin och zidovudin mot HIV. Ribavirin har i cellkultur visat sig hämma fosforylering av lamivudin, stavudin och zidovudin, vilket kan leda till minskad antiretroviral aktivitet. I en studie med ett annat pegylerat interferon i kombination med ribavirin observerades dock ingen farmakokinetisk (t.ex. plasmakoncentrationer eller intracellulära trifosforylerade aktiva metabolitkoncentrationer) eller farmakodynamisk (t.ex. förlust av HIV/HCV virologisk dämpning) interaktion när ribavirin och lamivudin (n) =18), stavudin (n=10) eller zidovudin (n=6) administrerades samtidigt som en del av en multidrug-regim hos HIV/HCV-infekterade patienter. Därför bör samtidig användning av ribavirin med något av dessa läkemedel användas med försiktighet.
Läkemedel som metaboliseras av Cytokrom P-450
Resultat av in vitro-studier med preparat av levermikrosomer från både människa och råtta indikerade liten eller ingen cytokrom P-450 enzymmedierad metabolism av ribavirin, med minimal potential för P-450 enzymbaserade läkemedelsinteraktioner.
Inga farmakokinetiska interaktioner noterades mellan INTRON A och REBETOL 200 mg kapslar i en farmakokinetisk multipeldosstudie.
Azatioprin
Användning av ribavirin för behandling av kronisk hepatit C hos patienter som får azatioprin har rapporterats inducera allvarlig pancytopeni och kan öka risken för azatioprinrelaterad myelotoxicitet. Inosinmonofosfatdehydrogenas (IMDH) krävs för en av azatioprins metaboliska vägar. Ribavirin är känt för att hämma IMDH, vilket leder till ackumulering av en azatioprinmetabolit, 6-metyltioinosinmonofosfat (6-MTITP), som är associerad med myelotoxicitet (neutropeni, trombocytopeni och anemi). Patienter som får azatioprin med ribavirin bör ha fullständiga blodvärden, inklusive trombocytantal, övervakas varje vecka under den första månaden, två gånger i månaden under den andra och tredje månaden av behandlingen, sedan en gång i månaden eller oftare om dos- eller annan behandlingsändring är nödvändig [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
VARNINGAR
Ingår som en del av FÖRSIKTIGHETSÅTGÄRDER sektion.
FÖRSIKTIGHETSÅTGÄRDER
Graviditet
REBETOL 200mg kapslar och oral lösning kan orsaka fosterskador och det ofödda barnets död. Behandling med REBETOL ska inte påbörjas förrän en rapport om ett negativt graviditetstest har erhållits omedelbart före planerad behandlingsstart. Patienter bör använda minst två former av preventivmedel och ha månatliga graviditetstest under behandlingen och under 6-månadersperioden efter att behandlingen har avbrutits. Extrem försiktighet måste iakttas för att undvika graviditet hos kvinnliga patienter och hos kvinnliga partner till manliga patienter. REBETOL 200mg har visat signifikanta teratogena och embryocidala effekter hos alla djurarter där adekvata studier har utförts. Dessa effekter inträffade vid doser som låg som en tjugondel av den rekommenderade humandosen av ribavirin. Behandling med REBETOL 200 mg ska inte påbörjas förrän ett negativt graviditetstest har rapporterats har erhållits omedelbart före planerad behandlingsstart [se LÅDA VARNING , KONTRAINDIKATIONER , Användning i specifika populationer , och PATIENTINFORMATION ].
Anemi
Den primära toxiciteten för ribavirin är hemolytisk anemi, som observerades hos cirka 10 % av REBETOL/INTRON A-behandlade försökspersoner i kliniska prövningar. Anemin i samband med REBETOL kapslar inträffar inom 1 till 2 veckor efter påbörjad behandling. Eftersom den initiala minskningen av hemoglobin kan vara signifikant, rekommenderas att hemoglobin eller hematokrit erhålls före behandlingsstart och vecka 2 och vecka 4 av behandlingen, eller oftare om det är kliniskt indicerat. Patienterna ska sedan följas om det är kliniskt lämpligt [se DOSERING OCH ADMINISTRERING ].
Fatala och icke-fatala hjärtinfarkter har rapporterats hos patienter med anemi orsakad av REBETOL. Patienter bör utvärderas med avseende på underliggande hjärtsjukdom innan behandling med ribavirin påbörjas. Patienter med redan existerande hjärtsjukdom bör få elektrokardiogram administrerat före behandling och bör övervakas på lämpligt sätt under behandlingen. Om det finns någon försämring av kardiovaskulär status ska behandlingen avbrytas eller avbrytas [se DOSERING OCH ADMINISTRERING ]. Eftersom hjärtsjukdom kan förvärras av läkemedelsinducerad anemi, bör patienter med en historia av signifikant eller instabil hjärtsjukdom inte använda REBETOL.
Pankreatit
Behandling med REBETOL och INTRON A eller PegIntron ska avbrytas hos patienter med tecken och symtom på pankreatit och avbrytas hos patienter med bekräftad pankreatit.
Lungsjukdomar
Lungsymtom, inklusive dyspné, lunginfiltrat, pneumonit, pulmonell hypertoni och lunginflammation, har rapporterats under behandling med REBETOL med kombinationsbehandling med alfa-interferon; enstaka fall av dödlig lunginflammation har inträffat. Dessutom har sarkoidos eller exacerbation av sarkoidos rapporterats. Om det finns tecken på lunginfiltrat eller lungfunktionsnedsättning ska patienten övervakas noggrant och om så är lämpligt ska kombinationsbehandlingen avbrytas.
Oftalmologiska störningar
Ribavirin används i kombinationsbehandling med alfa-interferoner. Minskad eller förlust av synen, retinopati inklusive makulaödem, retinal artär eller ven, trombos, retinala blödningar och bomullsfläckar, optikusneurit, papillödem och serös näthinneavlossning induceras eller förvärras av behandling med alfa-interferoner. Alla patienter bör genomgå en synundersökning vid baslinjen. Patienter med redan existerande oftalmologiska störningar (t.ex. diabetisk eller hypertensiv retinopati) bör genomgå periodiska oftalmologiska undersökningar under kombinationsbehandling med alfa-interferonbehandling. Varje patient som utvecklar ögonsymtom bör få en omedelbar och fullständig ögonundersökning. Kombinationsbehandling med alfa-interferoner bör avbrytas hos patienter som utvecklar nya eller förvärrade oftalmologiska störningar.
Laboratorietester
PegIntron i kombination med ribavirin kan orsaka allvarliga minskningar av antalet neutrofiler och trombocyter samt hematologiska, endokrina (t.ex. TSH) och leveravvikelser.
Patienter som får kombinationsbehandling med PegIntron/REBETOL bör genomgå hematologiska och blodkemiska tester innan behandlingen påbörjas och sedan regelbundet därefter. I den kliniska prövningen för vuxna mättes fullständigt blodvärde (inklusive hemoglobin-, neutrofil- och trombocytantal) och kemi (inklusive ASAT, ALT, bilirubin och urinsyra) under behandlingsperioden vid vecka 2, 4, 8, 12 och sedan med 6 veckors intervall, eller oftare om avvikelser utvecklades. Hos pediatriska försökspersoner utvärderades samma laboratorieparametrar med ytterligare bedömning av hemoglobin vid behandlingsvecka 6. TSH-nivåer mättes var 12:e vecka under behandlingsperioden. HCV-RNA bör mätas regelbundet under behandlingen [se DOSERING OCH ADMINISTRERING ].
Tand- och parodontala sjukdomar
Dentala och parodontala störningar har rapporterats hos patienter som får kombinationsbehandling med ribavirin och interferon eller peginterferon. Dessutom kan muntorrhet ha en skadlig effekt på tänder och slemhinnor i munnen under långtidsbehandling med kombinationen av REBETOL och pegylerat eller icke-pegylerat interferon alfa-2b. Patienter bör borsta tänderna noggrant två gånger dagligen och genomgå regelbundna tandundersökningar. Om kräkningar inträffar ska de rådas att skölja munnen noggrant efteråt.
Samtidig administrering av Azatioprin
Pancytopeni (markerad minskning av röda blodkroppar, neutrofiler och trombocyter) och benmärgssuppression har rapporterats i litteraturen som inträffar inom 3 till 7 veckor efter samtidig administrering av pegylerat interferon/ribavirin och azatioprin. Hos detta begränsade antal patienter (n=8) var myelotoxiciteten reversibel inom 4 till 6 veckor efter utsättande av både HCV antiviral behandling och samtidig azatioprin och återkom inte vid återinförande av någondera behandlingen ensam. PegIntron, REBETOL och azatioprin ska avbrytas för pancytopeni, och pegylerat interferon/ribavirin ska inte återinföras med samtidig azatioprin [se LÄKEMEDELSINTERAKTIONER ].
Påverkan på tillväxten - Pediatrisk användning
Data om effekterna av PegIntron och REBETOL på tillväxten kommer från en öppen studie på försökspersoner i åldern 3 till 17 år, där vikt- och längdförändringar jämförs med amerikanska normativa befolkningsdata. I allmänhet släpar vikt- och höjdökningen hos pediatriska patienter som behandlats med PegIntron och REBETOL 200 mg efter vad som förutspåtts av normativa populationsdata för hela behandlingens längd. Allvarligt hämmad tillväxthastighet (mindre än 3:e percentilen) observerades hos 70 % av försökspersonerna under behandling. Efter behandling inträffade återhämtningstillväxt och viktökning hos de flesta försökspersoner. Långtidsuppföljningsdata från pediatriska försökspersoner indikerar dock att PegIntron i kombinationsbehandling med REBETOL kan inducera en tillväxthämning som resulterar i minskad vuxenlängd hos vissa patienter [se NEGATIVA REAKTIONER ].
På liknande sätt sågs en påverkan på tillväxten hos försökspersoner efter behandling med REBETOL 200 mg och INTRON A kombinationsbehandling under ett år. I en långtidsuppföljningsstudie av ett begränsat antal av dessa försökspersoner, resulterade kombinationsbehandling i minskad slutlig vuxenlängd hos vissa försökspersoner [se NEGATIVA REAKTIONER ].
Användningsskydd
Baserat på resultat från kliniska prövningar är monoterapi med ribavirin inte effektivt för behandling av kronisk hepatit C-virusinfektion; därför får REBETOL 200 mg kapslar eller oral lösning inte användas ensamt. Säkerheten och effekten av REBETOL kapslar och oral lösning har endast fastställts när de används tillsammans med INTRON A eller PegIntron (inte andra interferoner) som kombinationsbehandling.
Säkerheten och effekten av behandling med REBETOL/INTRON A och PegIntron för behandling av HIV-infektion, adenovirus, RSV, parainfluensa eller influensainfektioner har inte fastställts. REBETOL 200mg kapslar ska inte användas för dessa indikationer. Ribavirin för inhalation har separat märkning, som bör konsulteras om inhalationsbehandling med ribavirin övervägs.
Det finns betydande biverkningar orsakade av behandling med REBETOL/INTRON A eller PegIntron, inklusive svår depression och självmordstankar, hemolytisk anemi, undertryckande av benmärgsfunktion, autoimmuna och infektionssjukdomar, lungdysfunktion, pankreatit och diabetes. Självmordstankar eller självmordsförsök förekom oftare bland pediatriska patienter, främst ungdomar, jämfört med vuxna patienter (2,4 % mot 1 %) under behandling och uppföljning utanför behandlingen. Märkning för INTRON A och PegIntron bör ses över i sin helhet för ytterligare säkerhetsinformation innan kombinationsbehandlingen påbörjas.
Information om patientrådgivning
Rekommendera patienten att läsa den FDA-godkända patientmärkningen ( Läkemedelsguide ).
Anemi
Den vanligaste biverkningen som inträffar med REBETOL kapslar är anemi, som kan vara allvarlig [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER och NEGATIVA REAKTIONER ]. Patienter bör informeras om att laboratorieutvärderingar krävs innan behandlingen påbörjas och med jämna mellanrum därefter [se DOSERING OCH ADMINISTRERING ]. Det rekommenderas att patienterna är väl hydrerade, särskilt under de inledande stadierna av behandlingen.
Graviditet
Patienterna måste informeras om att REBETOL 200 mg kapslar och oral lösning kan orsaka fosterskador och det ofödda barnets död. REBETOL 200mg får inte användas av kvinnor som är gravida eller av män vars kvinnliga partner är gravida. Extrem försiktighet måste iakttas för att undvika graviditet hos kvinnliga patienter och hos kvinnliga partner till manliga patienter som tar REBETOL. REBETOL ska inte påbörjas förrän en rapport om ett negativt graviditetstest har erhållits omedelbart innan behandlingen påbörjas. Patienterna måste utföra ett graviditetstest varje månad under behandlingen och under 6 månader efter behandlingen.
Kvinnor i fertil ålder måste rådas om användning av effektiva preventivmedel (två pålitliga former) innan behandlingen påbörjas. Patienter (män och kvinnor) måste informeras om de teratogena/embryocidala riskerna och måste instrueras att använda effektiv preventivmetod under REBETOL 200 mg och under 6 månader efter behandling. Patienter (män och kvinnor) bör rådas att omedelbart meddela läkaren i händelse av graviditet [se KONTRAINDIKATIONER , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , och Användning i specifika populationer ].
Om graviditet inträffar under behandling eller under 6 månader efter behandling, måste patienten informeras om den teratogena risken med REBETOL 200 mg-behandling för fostret. Patienter, eller partners till patienter, bör omedelbart rapportera varje graviditet som inträffar under behandlingen eller inom 6 månader efter avslutad behandling till sin läkare. Förskrivare bör rapportera sådana fall genom att ringa 1-800-593-2214.
Risker kontra fördelar
Patienter som får REBETOL-kapslar ska informeras om fördelarna och riskerna med behandlingen, inriktas på lämplig användning och remitteras till patienten. Läkemedelsguide . Patienterna bör informeras om att effekten av behandling av hepatit C-infektion på överföring inte är känd och att lämpliga försiktighetsåtgärder bör vidtas för att förhindra överföring av hepatit C-virus.
Patienter ska informeras om vad de ska göra om de missar en dos av REBETOL; den missade dosen ska tas så snart som möjligt under samma dag. Patienter ska inte dubbla nästa dos. Patienter bör rådas att kontakta sin vårdgivare om de har frågor.
Icke-klinisk toxikologi
Karcinogenes, Mutagenes, Nedsatt fertilitet
Carcinogenes
Ribavirin orsakade inte en ökning av någon tumörtyp när det administrerades under 6 månader i den transgena p53-deficienta musmodellen vid doser upp till 300 mg/kg (uppskattad human ekvivalent på 25 mg/kg baserat på kroppsytajustering för en vuxen på 60 kg cirka 1,9 gånger den maximala rekommenderade dagliga dosen för människor). Ribavirin var icke-karcinogent när det administrerades i 2 år till råttor i doser upp till 40 mg/kg (uppskattad humanekvivalent på 5,71 mg/kg baserat på justering av kroppsyta för en vuxen 60 kg).
Mutagenes
Ribavirin visade ökad incidens av mutationer och celltransformation i flera genotoxicitetsanalyser. Ribavirin var aktivt i Balb/3T3 in vitro celltransformationsanalysen. Mutagen aktivitet observerades i muslymfomanalysen och vid doser på 20 till 200 mg/kg (uppskattad human ekvivalent av 1,67 till 16,7 mg/kg, baserat på justering av kroppsyta för en vuxen 60 kg; 0,1 till 1 gånger det maximala rekommenderad human 24-timmarsdos av ribavirin) i en mikronukleusanalys på mus. En dominant dödlig analys på råttor var negativ, vilket indikerar att om mutationer inträffade hos råttor så överfördes de inte genom manliga könsceller.
Nedsatt fertilitet
Ribavirin visade signifikanta embryocidala och teratogena effekter vid doser långt under den rekommenderade humana dosen hos alla djurarter där adekvata studier har utförts. Missbildningar av skalle, gom, öga, käke, lemmar, skelett och mag-tarmkanalen noterades. Incidensen och svårighetsgraden av teratogena effekter ökade med eskalering av läkemedelsdosen. Överlevnaden för foster och avkommor minskade. I konventionella embryotoxicitets-/teratogenicitetsstudier på råttor och kaniner, var dosnivåerna utan effekt långt under de för föreslagen klinisk användning (0,3 mg/kg/dag för både råtta och kanin; cirka 0,06 gånger den rekommenderade humana 24-timmarsdosen av ribavirin). Ingen maternell toxicitet eller effekter på avkomma observerades i en peri/postnatal toxicitetsstudie på råttor som doserades oralt med upp till 1 mg/kg/dag (uppskattad human ekvivalentdos på 0,17 mg/kg baserat på justering av kroppsyta för en vuxen på 60 kg ; ungefär 0,01 gånger den maximala rekommenderade humana 24-timmarsdosen av ribavirin) [se KONTRAINDIKATIONER , och VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Fertila kvinnor och partners till fertila kvinnor ska inte få REBETOL om inte patienten och hans/hennes partner använder effektiv preventivmedel (två tillförlitliga former). Baserat på en halveringstid för flera doser (t1/2) av ribavirin på 12 dagar, måste effektiv preventivmetod användas i 6 månader efter behandlingen (t.ex. 15 halveringstider för clearance för ribavirin).
REBETOL ska användas med försiktighet till fertila män. I studier på möss för att utvärdera tidsförloppet och reversibiliteten av ribavirin-inducerad testikeldegeneration vid doser på 15 till 150 mg/kg/dag (uppskattad human ekvivalent av 1,25 till 12,5 mg/kg/dag, baserat på justering av kroppsyta för en 60 kg vuxen; 0,1-0,8 gånger den maximala humana 24-timmarsdosen av ribavirin) administrerat under 3 eller 6 månader, avvikelser i spermier inträffade. Efter avslutad behandling var i huvudsak total återhämtning från ribavirininducerad testikeltoxicitet uppenbar inom 1 eller 2 spermatogenescykler.
Användning i specifika populationer
Graviditet
Graviditetskategori X
[Ser KONTRAINDIKATIONER , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , och Icke-klinisk toxikologi ].
Behandling och efterbehandling:
Potentiell risk för fostret
Ribavirin är känt för att ackumuleras i intracellulära komponenter varifrån det elimineras mycket långsamt. Det är inte känt om ribavirin som finns i spermier kommer att utöva en potentiell teratogen effekt vid befruktning av ägget. I en studie på råttor drogs slutsatsen att dominant dödlighet inte inducerades av ribavirin vid doser upp till 200 mg/kg under 5 dagar (uppskattade humana ekvivalenta doser på 7,14 till 28,6 mg/kg, baserat på kroppsytajustering för 60 kg vuxen; upp till 1,7 gånger den maximala rekommenderade humandosen av ribavirin). På grund av de potentiella humana teratogena effekterna av ribavirin bör manliga patienter dock rådas att vidta alla försiktighetsåtgärder för att undvika risken för graviditet för sina kvinnliga partners.
Kvinnor i fertil ålder ska inte få REBETOL om de inte använder effektiv preventivmedel (två pålitliga former) under behandlingsperioden. Dessutom bör effektiv preventivmetod användas i 6 månader efter behandling baserat på en halveringstid för flera doser (t1/2) av ribavirin på 12 dagar.
Manliga patienter och deras kvinnliga partner måste använda effektiv preventivmetod (två tillförlitliga former) under behandling med REBETOL och under 6 månader efter terapiperioden (t.ex. 15 halveringstider för ribavirin-clearance från kroppen).
Ett ribavirin-graviditetsregister har upprättats för att övervaka mödernet-foster-utfall av graviditeter hos kvinnliga patienter och kvinnliga partners till manliga patienter som exponerats för ribavirin under behandlingen och i 6 månader efter avslutad behandling. Läkare och patienter uppmanas att rapportera sådana fall genom att ringa 1-800-593-2214.
Ammande mödrar
Det är inte känt om REBETOL-produkten utsöndras i modersmjölk. På grund av risken för allvarliga biverkningar från läkemedlet hos ammande spädbarn, bör ett beslut fattas om att avbryta amningen eller att skjuta upp eller avbryta behandlingen med REBETOL.
Pediatrisk användning
Säkerhet och effektivitet för REBETOL 200 mg i kombination med PegIntron har inte fastställts hos pediatriska patienter under 3 år. För behandling med REBETOL/INTRON A bör tecken på sjukdomsprogression, såsom leverinflammation och fibros, samt prognostiska faktorer för respons, HCV-genotyp och virusmängd beaktas vid beslut om att behandla en pediatrisk patient. Fördelarna med behandlingen bör vägas mot de observerade säkerhetsresultaten.
Långtidsuppföljningsdata från pediatriska försökspersoner indikerar att REBETOL i kombination med PegIntron eller med INTRON A kan inducera en tillväxthämning som resulterar i minskad längd hos vissa patienter [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER och NEGATIVA REAKTIONER ].
Självmordstankar eller självmordsförsök förekom oftare bland pediatriska patienter, främst ungdomar, jämfört med vuxna patienter (2,4 % mot 1 %) under behandling och uppföljning utanför behandlingen [ser VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. Som hos vuxna patienter upplevde pediatriska patienter andra psykiatriska biverkningar (t.ex. depression, emotionell labilitet, somnolens), anemi och neutropeni [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Geriatrisk användning
Kliniska prövningar av behandling med REBETOL/INTRON A eller PegIntron inkluderade inte tillräckligt många försökspersoner i åldern 65 år och äldre för att avgöra om de svarar annorlunda än yngre försökspersoner.
REBETOL 200mg är känt för att utsöndras till stor del via njurarna, och risken för toxiska reaktioner på detta läkemedel kan vara större hos patienter med nedsatt njurfunktion. Eftersom äldre patienter ofta har nedsatt njurfunktion bör man vara försiktig vid val av dos. Njurfunktionen bör övervakas och dosjusteringar bör göras i enlighet med detta. REBETOL 200 mg ska inte användas till patienter med kreatininclearance mindre än 50 ml/min [se KONTRAINDIKATIONER ].
Generellt sett bör REBETOL 200 mg kapslar administreras till äldre patienter med försiktighet, med början i den nedre delen av doseringsintervallet, vilket återspeglar den högre frekvensen av nedsatt lever- och hjärtfunktion, och av samtidig sjukdom eller annan läkemedelsbehandling. I kliniska prövningar hade äldre försökspersoner en högre frekvens av anemi (67 %) än yngre patienter (28 %) [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Organtransplantationsmottagare
Säkerheten och effekten av INTRON A och PegIntron enbart eller i kombination med REBETOL för behandling av hepatit C hos lever- eller andra organtransplanterade mottagare har inte fastställts. I ett litet (n=16) singelcenter, okontrollerat fall, var njursvikt hos njurtransplantatmottagare som fick interferon alfa och ribavirin kombinationsterapi mer frekvent än förväntat från centrets tidigare erfarenhet av njurtransplantatmottagare som inte fick kombinationsbehandling. Förhållandet mellan njursvikt och renal allotransplantatavstötning är inte klart.
HIV eller HBV saminfektion
Säkerheten och effekten av PegIntron/REBETOL 200 mg och INTRON A/REBETOL för behandling av patienter med HCV samtidigt infekterade med HIV eller HBV har inte fastställts.
ÖVERDOS
Det finns begränsad erfarenhet av överdosering. Akut intag av upp till 20 g REBETOL-kapslar, INTRON A-intag av upp till 120 miljoner enheter och subkutana doser av INTRON A upp till 10 gånger de rekommenderade doserna har rapporterats. Primära effekter som har observerats är ökad incidens och svårighetsgrad av biverkningar relaterade till terapeutisk användning av INTRON A och REBETOL. Leverenzymavvikelser, njursvikt, blödning och hjärtinfarkt har dock rapporterats vid administrering av enstaka subkutana doser av INTRON A som överstiger doseringsrekommendationerna.
Det finns ingen specifik motgift mot överdosering av INTRON A eller REBETOL 200 mg, och hemodialys och peritonealdialys är inte effektiva för behandling av överdosering av dessa medel.
KONTRAINDIKATIONER
REBETOL kombinationsterapi är kontraindicerat vid:
- kvinnor som är gravida. REBETOL 200mg kan orsaka fosterskador när det administreras till en gravid kvinna. REBETOL är kontraindicerat för kvinnor som är eller kan bli gravida. Om REBETOL används under graviditet, eller om patienten blir gravid medan du tar REBETOL 200 mg, ska patienten informeras om den potentiella faran för hennes foster [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , Användning i specifika populationer , och PATIENTINFORMATION ]
- män vars kvinnliga partner är gravida
- patienter med kända överkänslighetsreaktioner såsom Stevens-Johnsons syndrom, toxisk epidermal nekrolys och erythema multiforme mot ribavirin eller någon komponent i produkten
- patienter med autoimmun hepatit
- patienter med hemoglobinopati (t.ex. talassemi major, sicklecellanemi)
- patienter med kreatininclearance mindre än 50 ml/min. [ser Användning i specifika populationer och KLINISK FARMAKOLOGI ]
- Samtidig administrering av REBETOL och didanosin är kontraindicerat eftersom exponeringen för den aktiva metaboliten av didanosin (dideoxiadenosin 5'-trifosfat) ökar. Fatal leversvikt, såväl som perifer neuropati, pankreatit och symptomatisk hyperlaktatemi/laktacidos har rapporterats hos patienter som får didanosin i kombination med ribavirin [se LÄKEMEDELSINTERAKTIONER ].
KLINISK FARMAKOLOGI
Handlingsmekanism
Ribavirin är ett antiviralt medel [se Mikrobiologi ].
Farmakokinetik
Farmakokinetiska egenskaper hos engångs- och flerdoser hos vuxna sammanfattas i tabell 11. Ribavirin absorberades snabbt och omfattande efter oral administrering. Men på grund av first-pass metabolism var den absoluta biotillgängligheten i genomsnitt 64 % (44 %). Det fanns ett linjärt samband mellan dos och AUCtf (AUC från tidpunkt noll till sista mätbara koncentration) efter engångsdoser på 200 till 1200 mg ribavirin. Förhållandet mellan dos och Cmax var kurvlinjärt och tenderade att ha en asymptom över engångsdoser på 400 till 600 mg.
Vid upprepad oral dosering, baserat på AUC12h, observerades en 6-faldig ackumulering av ribavirin i plasma. Efter oral dosering med 600 mg två gånger dagligen uppnåddes steady-state efter cirka 4 veckor, med genomsnittliga steady-state plasmakoncentrationer på 2200 ng/ml (37%). Efter avbrytande av doseringen var den genomsnittliga halveringstiden 298 (30 %) timmar, vilket troligen återspeglar långsam eliminering från icke-plasmakompartment.
Effekt av antacida på absorption av ribavirin
Samtidig administrering av REBETOL-kapslar med ett antacidum innehållande magnesium, aluminium och simetikon resulterade i en 14 % minskning av den genomsnittliga AUCtf för ribavirin. Den kliniska relevansen av resultaten från denna endosstudie är okänd.
Vävnadsdistribution
Ribavirintransport in i icke-plasmakompartment har studerats mest omfattande i röda blodkroppar och har identifierats för att huvudsakligen ske via en ekvilibrativ nukleosidtransportör av es-typ. Denna typ av transportör finns på praktiskt taget alla celltyper och kan stå för den omfattande distributionsvolymen. Ribavirin binder inte till plasmaproteiner.
Metabolism och utsöndring
Ribavirin har två metabolismvägar: (i) en reversibel fosforyleringsväg i kärnförsedda celler; och (ii) en nedbrytningsväg som involverar deribosylering och amidhydrolys för att ge en triazolkarboxylsyrametabolit. Ribavirin och dess metaboliter av triazolkarboxamid och triazolkarboxylsyra utsöndras via njurarna. Efter oral administrering av 600 mg 14C-ribavirin eliminerades cirka 61 % och 12 % av radioaktiviteten i urinen respektive avföringen på 336 timmar. Oförändrat ribavirin stod för 17 % av den administrerade dosen.
Särskilda populationer
Renal dysfunktion
Farmakokinetiken för ribavirin utvärderades efter administrering av en oral engångsdos (400 mg) ribavirin till icke HCV-infekterade försökspersoner med olika grader av njurdysfunktion. Det genomsnittliga AUCtf-värdet var tre gånger högre hos försökspersoner med kreatininclearance-värden mellan 10 till 30 ml/min jämfört med kontrollpersoner (kreatininclearance större än 90 ml/min). Hos försökspersoner med kreatininclearance-värden mellan 30 och 60 ml/min var AUCtf två gånger högre jämfört med kontrollpersoner. Den ökade AUCtf verkar bero på minskning av renalt och icke-renalt clearance hos dessa patienter. Fas 3-effektstudier inkluderade försökspersoner med kreatininclearance-värden högre än 50 ml/min. Ribavirins farmakokinetik för flera doser kan inte exakt förutsägas hos patienter med nedsatt njurfunktion. Ribavirin avlägsnas inte effektivt genom hemodialys.
Patienter med kreatininclearance mindre än 50 ml/min ska inte behandlas med REBETOL [se KONTRAINDIKATIONER ].
Leverdysfunktion
Effekten av leverdysfunktion utvärderades efter en oral engångsdos av ribavirin (600 mg). De genomsnittliga AUCtf-värdena var inte signifikant olika hos försökspersoner med mild, måttlig eller svår leverdysfunktion (Child-Pugh-klassificering A, B eller C) jämfört med kontrollpersoner. De genomsnittliga Cmax-värdena ökade dock med svårighetsgraden av leverdysfunktion och var dubbelt så hög hos patienter med allvarlig leverdysfunktion jämfört med kontrollpersoner.
Äldre patienter
Farmakokinetiska utvärderingar hos äldre försökspersoner har inte utförts.
Kön
Det observerades inga kliniskt signifikanta farmakokinetiska skillnader i en endosstudie med 18 manliga och 18 kvinnliga försökspersoner.
Pediatriska patienter
Farmakokinetiska egenskaper med flera doser för REBETOL 200 mg kapslar och INTRON A hos pediatriska patienter med kronisk hepatit C mellan 5 och 16 år sammanfattas i tabell 12. Farmakokinetiken för REBETOL 200 mg och INTRON A (dosnormaliserade) är likartade hos vuxna och pediatriska ämnen.
Fullständiga farmakokinetiska egenskaper hos REBETOL oral lösning har inte fastställts hos pediatriska försökspersoner. Ribavirin Cmin-värden var liknande efter administrering av REBETOL oral lösning eller REBETOL 200 mg kapslar under 48 veckors behandling hos pediatriska försökspersoner (3 till 16 års ålder).
En klinisk prövning på pediatriska försökspersoner med kronisk hepatit C mellan 3 och 17 år utfördes där farmakokinetiken för PegIntron och REBETOL (kapslar och oral lösning) utvärderades. Hos pediatriska försökspersoner som fick kroppsyta-justerad dosering av PegIntron vid 60 mikrogram/m²/vecka, förutspåddes den logtransformerade kvoten för exponeringen under doseringsintervallet vara 58 % [90 % KI: 141 %, 177 %] högre än observerats hos vuxna som fick 1,5 mikrogram/kg/vecka. Farmakokinetiken för REBETOL (dosnormaliserad) i denna studie liknade den som rapporterades i en tidigare studie av REBETOL 200 mg i kombination med INTRON A hos pediatriska försökspersoner och hos vuxna.
Effekt av mat på absorption av ribavirin
Både AUCtf och Cmax ökade med 70 % när REBETOL-kapslar administrerades tillsammans med en måltid med hög fetthalt (841 kcal, 53,8 g fett, 31,6 g protein och 57,4 g kolhydrat) i en farmakokinetisk endosstudie [se DOSERING OCH ADMINISTRERING ].
Mikrobiologi
Handlingsmekanism
Mekanismen genom vilken ribavirin bidrar till dess antivirala effekt på kliniken är inte helt klarlagd. Ribavirin har direkt antiviral aktivitet i vävnadskultur mot många RNA-virus. Ribavirin ökar mutationsfrekvensen i arvsmassan hos flera virus och ribavirintrifosfat hämmar HCV-polymeras i en biokemisk reaktion.
Antiviral aktivitet i cellkultur
Den antivirala aktiviteten av ribavirin i HCV-replikonet är inte väl förstått och har inte definierats på grund av den cellulära toxiciteten av ribavirin. Direkt antiviral aktivitet har observerats i vävnadsodling av andra RNA-virus. Anti-HCV-aktiviteten hos interferon påvisades i cell som innehöll självreplikerande HCV-RNS (HCV-replikonceller) eller HCV-infektion.
Motstånd
HCV-genotyper visar stor variation i deras svar på pegylerad rekombinant human interferon/ribavirinterapi. Genetiska förändringar associerade med det variabla svaret har inte identifierats.
Korsmotstånd
Det finns ingen rapporterad korsresistens mellan pegylerade/icke-pegylerade interferoner och ribavirin.
Djurtoxikologi och farmakologi
Långtidsstudier på mus och råtta [18 till 24 månader; doser på 20 till 75 respektive 10 till 40 mg/kg/dag (uppskattade humana ekvivalentdoser på 1,67 till 6,25 respektive 1,43 till 5,71 mg/kg/dag, baserat på kroppsytajustering för en vuxen på 60 kg; ca. 0,1 till 0,4 gånger den maximala humana 24-timmarsdosen av ribavirin)] har visat ett samband mellan kronisk ribavirinexponering och ökad förekomst av vaskulära lesioner (mikroskopiska blödningar) hos möss. Hos råttor inträffade retinal degeneration hos kontroller, men incidensen ökade hos ribavirinbehandlade råttor.
en studie där råttungar doserades postnatalt med ribavirin i doser på 10, 25 och 50 mg/kg/dag, inträffade läkemedelsrelaterade dödsfall vid 50 mg/kg (vid plasmakoncentrationer av råttungar under humana plasmakoncentrationer på människa) terapeutisk dos) mellan studiedagarna 13 och 48. Råttungar doserade från dag 7 till 63 efter födseln visade en mindre dosrelaterad minskning av den totala tillväxten vid alla doser, vilket därefter manifesterades som en liten minskning av kroppsvikt, längd på krona och bakdel, och benlängd. Dessa effekter visade tecken på reversibilitet och inga histopatologiska effekter på ben observerades. Inga effekter av ribavirin observerades avseende neurobeteende eller reproduktiv utveckling.
Kliniska studier
Klinisk studie 1 utvärderade PegIntron monoterapi. Ser PegIntron-märkning för information om denna prövning.
REBETOL/PegIntron kombinationsterapi
Vuxna ämnen
Studie 2
En randomiserad studie jämförde behandlingen med två PegIntron/REBETOL 200 mg-regimer [PegIntron 1,5 mcg/kg subkutant en gång i veckan/REBETOL 800 mg oralt dagligen (uppdelade doser); PegIntron 1,5 mcg/kg subkutant en gång i veckan i 4 veckor sedan 0,5 mcg/kg subkutant en gång i veckan i 44 veckor/REBETOL 1000 eller 1200 mg oralt dagligen (i uppdelade doser)] med INTRON A [3 MIE subkutant tre gånger i veckan/REBET eller REBET 1200 mg oralt dagligen (i uppdelade doser)] hos 1530 vuxna med kronisk hepatit C. Interferonnaiva försökspersoner behandlades i 48 veckor och följdes under 24 veckor efter behandling. Kvalificerade försökspersoner hade kompenserad leversjukdom, detekterbart HCV-RNA, förhöjd ALT och leverhistopatologi i överensstämmelse med kronisk hepatit.
Svar på behandling definierades som odetekterbart HCV-RNA 24 veckor efter behandling (se tabell 13). Svarsfrekvensen på dosen PegIntron 1,5 mcg/kg och ribavirin 800 mg var högre än svarsfrekvensen på INTRON A/REBETOL (se tabell 13). Svarsfrekvensen för PegIntron 1,5→0,5 mcg/kg/REBETOL 200 mg var i stort sett densamma som svar på INTRON A/REBETOL (data visas inte).
Patienter med viral genotyp 1, oavsett virusmängd, hade en lägre svarsfrekvens på PegIntron (1,5 mcg/kg)/REBETOL (800 mg) jämfört med försökspersoner med andra virala genotyper. Försökspersoner med både dåliga prognostiska faktorer (genotyp 1 och hög virusmängd) hade en svarsfrekvens på 30 % (78/256) jämfört med en svarsfrekvens på 29 % (71/247) med INTRON A/REBETOL kombinationsbehandling.
Försökspersoner med lägre kroppsvikt tenderade att ha högre biverkningsfrekvenser [se NEGATIVA REAKTIONER och högre svarsfrekvens än försökspersoner med högre kroppsvikt. Skillnader i svarsfrekvens mellan behandlingsarmarna varierade inte väsentligt med kroppsvikten.
Behandlingssvarsfrekvensen med PegIntron/REBETOL kombinationsbehandling var 49 % hos män och 56 % hos kvinnor. Svarsfrekvensen var lägre hos afroamerikanska och latinamerikanska personer och högre hos asiater jämfört med kaukasier. Även om afroamerikaner hade en högre andel dåliga prognostiska faktorer jämfört med kaukasier, var antalet studerade icke-kaukasier (11% av totalen) otillräckligt för att tillåta meningsfulla slutsatser om skillnader i svarsfrekvens efter justering för prognostiska faktorer i denna studie.
Leverbiopsier erhölls före och efter behandling hos 68 % av försökspersonerna. Jämfört med baslinjen observerades ungefär två tredjedelar av försökspersonerna i alla behandlingsgrupper ha en blygsam minskning av inflammation.
Studie 3
en stor samhällsbaserad studie i USA randomiserades 4913 försökspersoner med kronisk hepatit C till att få PegIntron 1,5 mikrogram/kg subkutant en gång i veckan i kombination med en REBETOL 200 mg dos på 800 till 1400 mg (viktbaserad dosering [WBD]) eller 800 mg (platt) oralt dagligen (i uppdelade doser) i 24 eller 48 veckor baserat på genotyp. Svar på behandling definierades som odetekterbart HCV-RNA (baserat på en analys med en nedre detektionsgräns på 125 IE/ml) 24 veckor efter behandling.
Behandling med PegIntron 1,5 mcg/kg och REBETOL 800 till 1400 mg resulterade i ett högre ihållande virologiskt svar jämfört med PegIntron i kombination med en platt 800 mg daglig dos av REBETOL. Försökspersoner som vägde mer än 105 kg fick den största fördelen med WBD, även om en blygsam fördel också observerades hos försökspersoner som vägde mer än 85 till 105 kg (se tabell 14). Fördelarna med WBD hos försökspersoner som väger mer än 85 kg observerades med HCV-genotyper 1-3. Det fanns inte tillräckligt med data för att dra slutsatser om andra genotyper. Användning av WBD resulterade i en ökad förekomst av anemi [se NEGATIVA REAKTIONER ].
Totalt 1552 försökspersoner som vägde mer än 65 kg i studie 3 hade genotyp 2 eller 3 och randomiserades till 24 eller 48 veckors behandling. Ingen ytterligare fördel observerades med den längre behandlingstiden.
Studie 4
En stor randomiserad studie jämförde säkerheten och effekten av behandling under 48 veckor med två PegIntron/REBETOL 200 mg regimer [PegIntron 1,5 mcg/kg och 1 mcg/kg subkutant en gång i veckan, båda i kombination med REBETOL 800 till 1400 mg PO dagligen (delat på två delar) doser)] och Pegasys 180 mikrogram subkutant en gång i veckan i kombination med Copegus 1000 till 1200 mg PO dagligen (i två uppdelade doser) hos 3070 behandlingsnaiva vuxna med kronisk hepatit C genotyp 1. I denna studie, avsaknad av tidig virologisk respons (ej detekterbar) HCV-RNA eller mer än eller lika med 2 log10-reduktion från baslinjen) vid behandling Vecka 12 var kriteriet för att avbryta behandlingen. SVR definierades som odetekterbart HCV-RNA (Roche COBAS TaqMan-analys, en nedre kvantifieringsgräns på 27 IE/ml) 24 veckor efter behandling (se tabell 15).
Den totala SVR-frekvensen var likartad bland de tre behandlingsgrupperna. Oavsett behandlingsgrupp var SVR-frekvensen lägre hos patienter med dåliga prognostiska faktorer. Försökspersoner med dåliga prognostiska faktorer randomiserade till PegIntron (1,5 mcg/kg)/REBETOL eller Pegasys/Copegus uppnådde dock högre SVR-frekvenser jämfört med liknande försökspersoner som randomiserades till PegIntron 1 mikrogram/kg/REBETOL. För dosen PegIntron 1,5 mcg/kg och REBETOL var SVR-frekvensen för försökspersoner med och utan följande prognostiska faktorer som följer: skrumplever (10 % vs. 42 %), normala ALAT-nivåer (32 % vs. 42 %), viral grundlinje belastning större än 600 000 IE/ml (35 % vs. 61 %), 40 år och äldre (38 % vs. 50 %) och afroamerikansk ras (23 % vs. 44 %). Hos försökspersoner med odetekterbart HCV-RNA vid behandlingsvecka 12 som fick PegIntron (1,5 mcg/kg)/REBETOL 200 mg, var SVR-frekvensen 81 % (328/407).
Studie 5 - REBETOL/PegIntron kombinationsterapi vid tidigare behandlingsmisslyckanden
en icke-jämförande studie återbehandlades 2293 försökspersoner med måttlig till svår fibros som misslyckats med tidigare behandling med kombinationen alfa-interferon/ribavirin med PegIntron, 1,5 mikrogram/kg subkutant, en gång i veckan, i kombination med viktjusterat ribavirin. Kvalificerade försökspersoner inkluderade tidigare icke-svarare (försökspersoner som var HCV-RNA-positiva i slutet av minst 12 veckors behandling) och tidigare återfall (försökspersoner som var HCVRNA-negativa i slutet av minst 12 veckors behandling och därefter återfall efter efterbehandlingen uppföljning). Försökspersoner som var negativa vid vecka 12 behandlades i 48 veckor och följdes i 24 veckor efter behandling. Svar på behandling definierades som odetekterbart HCV-RNA 24 veckor efter behandling (mätt med ett forskningsbaserat test, detektionsgräns 125 IE/ml). Den totala svarsfrekvensen var 22 % (497/2293) (99 % KI: 19,5, 23,9). Patienter med följande egenskaper hade mindre sannolikhet att dra nytta av ombehandling: tidigare utebliven respons, tidigare behandling med pegylerat interferon, signifikant överbryggande fibros eller cirros och genotyp 1-infektion.
De ihållande virologiska svarsfrekvenserna för återbehandlingen efter baslinjeegenskaper sammanfattas i tabell 16.
Uppnåendet av ett odetekterbart HCV-RNA vid behandlingsvecka 12 var en stark prediktor för SVR. I denna studie uppnådde 1470 (64 %) försökspersoner inte ett odetekterbart HCV-RNA vid behandlingsvecka 12 och erbjöds att delta i långtidsbehandlingsstudier på grund av ett otillräckligt behandlingssvar. Av de 823 (36 %) försökspersonerna som inte kunde upptäckas med HCV-RNA vid behandlingsvecka 12, hade de infekterade med genotyp 1 en SVR på 48 % (245/507), med ett antal svar enligt fibrospoäng (F4-F2) av 39-55%. Försökspersoner infekterade med genotyp 2/3 som var HCV-RNA odetekterbara vid behandlingsvecka 12 hade en total SVR på 70 % (196/281), med ett antal svar genom fibrospoäng (F4-F2) på 60-83 %. För alla genotyper var högre fibrospoäng associerade med en minskad sannolikhet för att uppnå SVR.
Pediatriska ämnen
Tidigare obehandlade pediatriska försökspersoner i åldern 3 till 17 år med kompenserad kronisk hepatit C och detekterbart HCV-RNA behandlades med REBETOL 15 mg/kg per dag och PegIntron 60 mcg/m² en gång i veckan i 24 eller 48 veckor baserat på HCV-genotyp och baslinjevirus ladda. Alla försökspersoner skulle följas under 24 veckor efter behandling. Totalt 107 försökspersoner fick behandling, varav 52% var kvinnor, 89% var kaukasiska och 67% var infekterade med HCV genotyp 1. Försökspersoner infekterade med genotyp 1, 4 eller genotyp 3 med HCV-RNA större än eller lika med 600 000 IE/ml fick 48 veckors behandling medan de som var infekterade med genotyp 2 eller genotyp 3 med HCV-RNA mindre än 600 000 IE/ml fick 24 veckors behandling. Försöksresultaten sammanfattas i tabell 17.
REBETOL/INTRON En kombinationsterapi
Vuxna ämnen
Tidigare obehandlade ämnen
Vuxna med kompenserad kronisk hepatit C och detekterbart HCV-RNA (bedömd av ett centralt laboratorium med en forskningsbaserad RT-PCR-analys) som tidigare inte behandlats med alfa-interferonterapi inkluderades i två multicenter, dubbelblinda studier (USA och internationella) och randomiserades till att få REBETOL 200 mg kapslar 1200 mg/dag (1000 mg/dag för försökspersoner som väger mindre än eller lika med 75 kg) och INTRON A 3 MIE tre gånger i veckan eller INTRON A och placebo i 24 eller 48 veckor följt av 24 veckors uppföljning utanför terapin. Den internationella studien innehöll inte en 24-veckors INTRON A- och placebobehandlingsarm. Den amerikanska studien inkluderade 912 försökspersoner som vid baslinjen var 67 % män, 89 % kaukasiska med ett genomsnittligt Knodell HAI-poäng (I+II+III) på 7,5 och 72 % genotyp 1. Den internationella studien, genomförd i Europa, Israel , Kanada och Australien, inkluderade 799 försökspersoner (65 % män, 95 % kaukasiska, genomsnittliga Knodell-poäng 6,8 och 58 % genotyp 1).
Försöksresultaten sammanfattas i tabell 18.
Av försökspersoner som inte hade uppnått HCV-RNA under detektionsgränsen för den forskningsbaserade analysen senast vecka 24 av REBETOL/INTRON A-behandling, svarade mindre än 5 % på ytterligare 24 veckors kombinationsbehandling.
Bland försökspersoner med HCV genotyp 1 behandlade med REBETOL/INTRON A-terapi som uppnådde HCV-RNA under detektionsgränsen för den forskningsbaserade analysen med 24 veckor, hade de som randomiserades till 48 veckors behandling högre virologiska svar jämfört med de i 24- veckas behandlingsgrupp. Det observerades ingen ökning av svarsfrekvensen för försökspersoner med HCV icke-genotyp 1 randomiserade till REBETOL/INTRON A-behandling under 48 veckor jämfört med 24 veckor.
Återfallsämnen
Försökspersoner med kompenserad kronisk hepatit C och detekterbart HCV-RNA (bedömd av ett centrallaboratorium med en forskningsbaserad RT-PCR-analys) som hade återfall efter en eller två kurer av interferonterapi (definierad som onormala serum-ALAT-nivåer) inkluderades i två multicenter, dubbelblinda studier (USA och internationella) och randomiserade för att få REBETOL 1200 mg/dag (1000 mg/dag för försökspersoner som väger ≤ 75 kg) och INTRON A 3 MIE tre gånger i veckan eller INTRON A och placebo i 24 veckor följt av 24 veckors uppföljning utanför behandlingen. Den amerikanska studien inkluderade 153 försökspersoner som vid baslinjen var 67 % män, 92 % kaukasiska med ett genomsnittligt Knodell HAI-poäng (I+II+III) på 6,8 och 58 % genotyp 1. Den internationella studien, genomförd i Europa, Israel , Kanada och Australien, inkluderade 192 försökspersoner (64 % män, 95 % kaukasiska, genomsnittlig Knodell-poäng 6,6 och 56 % genotyp 1). Försöksresultaten sammanfattas i Tabell 19.
Virologiska och histologiska svar var likartade bland manliga och kvinnliga försökspersoner i både de tidigare obehandlade och skovförsöken.
Pediatriska ämnen
Pediatriska försökspersoner i åldern 3 till 16 år med kompenserad kronisk hepatit C och detekterbart HCV-RNA (bedömd av ett centralt laboratorium med en forskningsbaserad RT-PCR-analys) behandlades med REBETOL 15 mg/kg per dag och INTRON A 3 MIE/ m² tre gånger i veckan i 48 veckor följt av 24 veckors uppföljning utanför behandlingen. Totalt 118 försökspersoner fick behandling, varav 57 % var män, 80 % kaukasiska och 78 % av genotyp 1. Försökspersoner yngre än 5 år fick REBETOL oral lösning och de som var 5 år eller äldre fick antingen REBETOL oral lösning eller kapslar.
Försöksresultaten sammanfattas i tabell 20.
Försökspersoner med viral genotyp 1, oavsett virusmängd, hade en lägre svarsfrekvens på INTRON A/REBETOL kombinationsbehandling jämfört med försökspersoner med genotyp icke-1, 36 % jämfört med 81 %. Försökspersoner med både dåliga prognostiska faktorer (genotyp 1 och hög virusmängd) hade en svarsfrekvens på 26 % (13/50).
PATIENTINFORMATION
Ingen information lämnas. Vänligen se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER sektion.