Epivir 150mg Lamivudine Användning, biverkningar och dosering. Pris i onlineapotek. Generiska läkemedel utan recept.
Vad är Epivir och hur används det?
Epivir 150 mg är ett receptbelagt läkemedel som används för att behandla symtom på HIV-infektion och kronisk hepatit B. Epivir 150 mg kan användas ensamt eller tillsammans med andra läkemedel.
Epivir tillhör en klass av läkemedel som kallas hepatit B/hepatit C-medel; HIV, NRTI.
Det är inte känt om Epivir är säkert och effektivt hos barn under 3 månaders ålder.
Vilka är de möjliga biverkningarna av Epivir 150 mg?
Epivir kan orsaka allvarliga biverkningar inklusive:
- nässelfeber,
- svårt att andas,
- svullnad av ansikte, läppar, tunga eller svalg,
- ovanlig muskelsmärta,
- magont,
- kräkningar,
- oregelbunden hjärtfrekvens,
- yrsel,
- känns kallt,
- svaghet,
- trötthet,
- svår smärta i övre delen av magen sprider sig till ryggen,
- illamående,
- snabb puls,
- svullnad runt mittpartiet,
- högersidiga övre magsmärtor,
- aptitlöshet,
- mörk urin,
- lerfärgad avföring,
- gulfärgning av huden eller ögonen (gulsot),
- feber,
- nattsvettningar,
- svullna körtlar,
- munsår,
- hosta,
- väsande andning,
- diarre,
- viktminskning,
- problem med att tala eller svälja,
- problem med balans eller ögonrörelser,
- taggig känsla,
- svullnad i nacken eller halsen (förstorad sköldkörtel),
- menstruationsförändringar, och
- impotens
Få medicinsk hjälp omedelbart om du har något av symtomen som anges ovan.
De vanligaste biverkningarna av Epivir inkluderar:
- illamående,
- diarre,
- huvudvärk,
- feber,
- trötthet,
- allmän dålig känsla,
- öronvärk eller full känsla,
- svårt att höra,
- dränering från örat,
- krångel hos ett barn,
- Täppt i näsan,
- nysning,
- ont i halsen, och
- hosta
Tala om för läkaren om du har någon biverkning som stör dig eller som inte försvinner.
Dessa är inte alla möjliga biverkningar av Epivir. För mer information, fråga din läkare eller apotekspersonal.
Ring din läkare för medicinsk rådgivning om biverkningar. Du kan rapportera biverkningar till FDA på 1-800-FDA-1088.
VARNING
EXACERBATIONER AV HEPATIT B, och OLIKA FORMULERINGAR AV EPIVIR.
Exacerbationer av hepatit B
Allvarliga akuta exacerbationer av hepatit B har rapporterats hos patienter som samtidigt är infekterade med hepatit B-virus (HBV) och humant immunbristvirus (HIV-1) och som har avbrutit behandlingen med EPIVIR. Leverfunktionen bör övervakas noggrant med både klinisk uppföljning och laboratorieuppföljning under minst flera månader hos patienter som avbryter behandlingen med EPIVIR och samtidigt är infekterade med HIV-1 och HBV. Om så är lämpligt kan initiering av antihepatit B-behandling vara motiverad [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER].
Viktiga skillnader mellan produkter som innehåller lamivudin
EPIVIR 150 mg tabletter och oral lösning (används för att behandla HIV-1-infektion) innehåller en högre dos av den aktiva ingrediensen (lamivudin) än EPIVIR-HBV tabletter och oral lösning (används för att behandla kronisk HBV-infektion). Patienter med HIV-1-infektion ska endast få doseringsformer som är lämpliga för behandling av HIV-1 [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER].
BESKRIVNING
EPIVIR (även känd som 3TC) är ett varumärke för lamivudin, en syntetisk nukleosidanalog med aktivitet mot HIV-1 och HBV. Det kemiska namnet på lamivudin är (2R,cis)-4-amino-1(2-hydroximetyl-1,3-oxatiolan-5-yl)-(1H)-pyrimidin-2-on. Lamivudin är (-)enantiomeren av en dideoxianalog av cytidin. Lamivudin har även kallats (-)2',3'-dideoxi, 3'tiacytidin. Den har en molekylformel av C8H11N3O3S och en molekylvikt på 229,3 g per mol. Den har följande strukturformel:
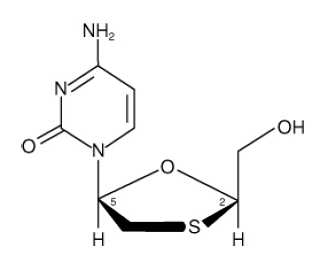
Lamivudin är ett vitt till benvitt kristallint fast ämne med en löslighet på cirka 70 mg per ml i vatten vid 20°C.
EPIVIR 150 mg tabletter är för oral administrering. Varje skårad 150 mg filmdragerad tablett innehåller 150 mg lamivudin och de inaktiva ingredienserna hypromellos, magnesiumstearat, mikrokristallin cellulosa, polyetylenglykol, polysorbat 80, natriumstärkelseglykolat och titandioxid.
Varje 300 mg filmdragerad tablett innehåller 300 mg lamivudin och de inaktiva ingredienserna svart järnoxid, hypromellos, magnesiumstearat, mikrokristallin cellulosa, polyetylenglykol, polysorbat 80, natriumstärkelseglykolat och titandioxid.
EPIVIR oral lösning är för oral administrering. En milliliter (1 ml) EPIVIR oral lösning innehåller 10 mg lamivudin (10 mg per ml) i en vattenlösning och de inaktiva ingredienserna konstgjorda jordgubbs- och bananaromer, citronsyra (vattenfri), metylparaben, propylenglykol, propylparaben, natriumcitrat (dihydrat) och sackaros (200 mg).
INDIKATIONER
EPIVIR 150 mg är en nukleosidanalog indicerad i kombination med andra antiretrovirala medel för behandling av humant immunbristvirus typ 1 (HIV-1)-infektion.
Användningsbegränsningar
- Doseringen av denna produkt är för HIV-1 och inte för HBV.
DOSERING OCH ADMINISTRERING
Rekommenderad dosering för vuxna patienter
Den rekommenderade dosen av EPIVIR 150 mg till HIV-1-infekterade vuxna är 300 mg dagligen, administrerad som antingen 150 mg oralt två gånger dagligen eller 300 mg oralt en gång dagligen med eller utan mat. Om lamivudin administreras till en patient som är infekterad med HIV-1 och HBV, bör den dos som indikeras för HIV-1-behandling användas som en del av en lämplig kombinationsregim [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Rekommenderad dosering för pediatriska patienter
EPIVIR 150 mg skårad tablett är den föredragna formuleringen för HIV-1-infekterade pediatriska patienter som väger minst 14 kg och för vilka en fast doseringsform är lämplig. Innan EPIVIR tabletter med skåror förskrivs, bör pediatriska patienter utvärderas med avseende på förmågan att svälja tabletter. För patienter som inte kan svälja EPIVIR 150 mg tabletter på ett säkert och tillförlitligt sätt, kan oral lösning förskrivas [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. Den rekommenderade orala dosen av EPIVIR 150 mg tabletter för HIV-1-infekterade pediatriska patienter presenteras i Tabell 1.
Oral lösning
Den rekommenderade dosen av EPIVIR 150 mg oral lösning till HIV-1-infekterade pediatriska patienter i åldern 3 månader och äldre är 5 mg per kg oralt två gånger dagligen eller 10 mg per kg oralt en gång dagligen (upp till maximalt 300 mg dagligen). administreras i kombination med andra antiretrovirala medel [se KLINISK FARMAKOLOGI ]. Tänk på HIV-1 virusmängd och CD4+ cellantal/procentandel när du väljer doseringsintervall för patienter som påbörjar behandling med oral lösning [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , KLINISK FARMAKOLOGI ].
Patienter med nedsatt njurfunktion
Doseringen av EPIVIR 150 mg justeras i enlighet med njurfunktionen. Dosjusteringar listas i Tabell 2 [se KLINISK FARMAKOLOGI ].
Ingen ytterligare dosering av EPIVIR 150 mg krävs efter rutinmässig (4 timmar) hemodialys eller peritonealdialys.
Även om det inte finns tillräckliga data för att rekommendera en specifik dosjustering av EPIVIR till pediatriska patienter med nedsatt njurfunktion, bör en minskning av dosen och/eller en ökning av doseringsintervallet övervägas.
HUR LEVERERAS
Doseringsformer och styrkor
EPIVIR Poängsatta tabletter
EPIVIR tabletter med skåror innehåller 150 mg lamivudin. Tabletterna är vita, diamantformade, skårade, filmdragerade tabletter präglade med ”GX CJ7” på båda sidor.
EPIVIR tabletter
EPIVIR tabletter innehåller 300 mg lamivudin. Tabletterna är grå, modifierade diamantformade, filmdragerade och graverade med ”GX EJ7” på ena sidan och släta på baksidan.
EPIVIR oral lösning
EPIVIR oral lösning innehåller 10 mg lamivudin per 1 ml. Lösningen är en klar, färglös till ljusgul vätska med jordgubbs-banansmak.
Förvaring Och Hantering
EPIVIR skårade tabletter innehåller 150 mg lamivudin, är vita, diamantformade, filmdragerade och präglade med "GX CJ7" på båda sidor. Förpackad enligt följande:
Flaska med 60 tabletter ( NDC 49702-203-18) med barnsäker stängning.
EPIVIR tabletter innehåller 300 mg lamivudin, är grå, modifierade diamantformade, filmdragerade och märkta med ”GX EJ7” på ena sidan och släta på baksidan. Förpackad enligt följande:
Flaska med 30 tabletter ( NDC 49702-204-13) med barnsäker stängning.
Rekommenderad förvaring
Förvara EPIVIR-tabletter vid 25°C (77°F); utflykter tillåtna till 15° till 30°C (59° till 86°F) [se USP-kontrollerad rumstemperatur ].
EPIVIR oral lösning är en klar, färglös till ljusgul vätska med jordgubbsbanansmak. Varje ml av lösningen innehåller 10 mg lamivudin. Förpackad enligt följande:
Plastflaska på 240 ml (NDC 49702-205-48) med barnsäker stängning. Denna produkt kräver inte beredning.
Förvaras i tättslutna flaskor vid 25°C (77°F) [se USP-kontrollerad rumstemperatur ].
Tillverkad för: ViiV Healthcare Research Triangle Park, NC 27709. av: GlaxoSmithKline Research Triangle Park, NC 27709, Tillverkad enligt överenskommelse från Shire Pharmaceuticals Group plc. Reviderad: april 2018
BIEFFEKTER
Följande biverkningar diskuteras i andra avsnitt av märkningen:
- Exacerbationer av hepatit B [se LÅDA VARNING , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
- Laktacidos och svår hepatomegali med steatos [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
- Leverdekompensation hos patienter som samtidigt är infekterade med HIV-1 och hepatit C [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
- Pankreatit [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
- Immunrekonstitutionssyndrom [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Erfarenhet av kliniska prövningar
Erfarenhet av kliniska prövningar i vuxna ämnen
Eftersom kliniska prövningar genomförs under vitt skilda förhållanden, kan biverkningsfrekvenser som observerats i de kliniska prövningarna av ett läkemedel inte direkt jämföras med frekvenser i de kliniska prövningarna av ett annat läkemedel och återspeglar kanske inte de frekvenser som observerats i klinisk praxis.
Säkerhetsprofilen för EPIVIR 150 mg hos vuxna är primärt baserad på 3 568 HIV-1-infekterade försökspersoner i 7 kliniska prövningar.
De vanligaste biverkningarna är huvudvärk, illamående, sjukdomskänsla, trötthet, nasala tecken och symtom, diarré och hosta.
Utvalda kliniska biverkningar hos mer än eller lika med 5 % av försökspersonerna under behandling med EPIVIR 150 mg två gånger dagligen plus RETROVIR 200 mg 3 gånger dagligen i upp till 24 veckor listas i Tabell 3.
Pankreatit
Pankreatit observerades hos 9 av 2 613 vuxna försökspersoner (0,3 %) som fick EPIVIR 150 mg i kontrollerade kliniska prövningar EPV20001, NUCA3001, NUCB3001, NUCA3002, NUCB3002 och NUCB3007 [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
EPIVIR 300 mg en gång dagligen
Typerna och frekvenserna av kliniska biverkningar som rapporterats hos försökspersoner som fick EPIVIR 300 mg en gång dagligen eller EPIVIR 150 mg två gånger dagligen (i 3-läkemedelskombinationsregimer i EPV20001 och EPV40001) under 48 veckor var likartade.
Utvalda laboratorieavvikelser som observerats under behandlingen sammanfattas i tabell 4.
Frekvensen av utvalda laboratorieavvikelser som rapporterades hos försökspersoner som fick EPIVIR 300 mg en gång dagligen eller EPIVIR 150 mg två gånger dagligen (i 3-läkemedelskombinationsregimer i EPV20001 och EPV40001) var likartade.
Erfarenhet av kliniska prövningar i pediatriska ämnen
EPIVIR oral lösning har studerats på 638 pediatriska försökspersoner i åldern 3 månader till 18 år i 3 kliniska prövningar.
Utvalda kliniska biverkningar och fysiska fynd med en frekvens som är större än eller lika med 5 % under behandling med EPIVIR 4 mg per kg två gånger dagligen plus RETROVIR 160 mg per m² 3 gånger dagligen vid behandlingsnaiva (mindre än eller lika med 56 dagars antiretroviral behandling) terapi) pediatriska ämnen listas i tabell 5.
Pankreatit
Pankreatit, som har varit dödlig i vissa fall, har observerats hos antiretrovirala nukleosid-erfarna pediatriska patienter som fått EPIVIR enbart eller i kombination med andra antiretrovirala medel. I en öppen dosupptrappningsstudie (NUCA2002) utvecklade 14 försökspersoner (14%) pankreatit medan de fick monoterapi med EPIVIR. Tre av dessa försökspersoner dog av komplikationer av pankreatit. I en andra öppen studie (NUCA2005) utvecklade 12 försökspersoner (18%) pankreatit. I studien ACTG300 observerades inte pankreatit hos 236 försökspersoner randomiserade till EPIVIR 150 mg plus RETROVIR. Pankreatit observerades hos 1 försöksperson i denna studie som fick öppen EPIVIR i kombination med RETROVIR och ritonavir efter utsättning av didanosinmonoterapi [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Parestesier och perifera neuropatier
Parestesier och perifera neuropatier rapporterades hos 15 försökspersoner (15 %) i försök NUCA2002, 6 försökspersoner (9 %) i försök NUCA2005 och 2 försökspersoner (mindre än 1 %) i försök med ACTG300.
Utvalda laboratorieavvikelser som upplevts av terapinaiva (mindre än eller lika med 56 dagars antiretroviral behandling) pediatriska patienter listas i Tabell 6.
Pediatriska försökspersoner en gång dagligen versus dosering två gånger dagligen (COL105677)
Säkerheten vid dosering en gång dagligen jämfört med dosering två gånger dagligen av EPIVIR 150 mg bedömdes i ARROW-studien. Den primära säkerhetsbedömningen i ARROW-studien baserades på biverkningar av grad 3 och grad 4. Frekvensen av biverkningar av grad 3 och 4 var liknande bland försökspersoner som randomiserades till dosering en gång dagligen jämfört med försökspersoner som randomiserades till dosering två gånger dagligen. En händelse av hepatit av grad 4 i kohorten en gång dagligen ansågs som osäker orsakssamband av prövaren och alla andra biverkningar av grad 3 eller 4 ansågs inte relaterade av prövaren.
Nyfödda
Begränsad korttidssäkerhetsinformation finns tillgänglig från 2 små okontrollerade studier i Sydafrika på nyfödda som får lamivudin med eller utan zidovudin under den första levnadsveckan efter moderns behandling med början vid vecka 38 eller 36 av graviditeten [se KLINISK FARMAKOLOGI ]. Utvalda biverkningar som rapporterats hos dessa nyfödda inkluderade ökade leverfunktionstester, anemi, diarré, elektrolytrubbningar, hypoglykemi, gulsot och hepatomegali, hudutslag, luftvägsinfektioner och sepsis; 3 nyfödda dog (1 av gastroenterit med acidos och kramper, 1 av traumatisk skada och 1 av okänd orsak). Två andra icke-dödliga fall av gastroenterit eller diarré rapporterades, inklusive 1 med konvulsioner; 1 spädbarn hade övergående njurinsufficiens i samband med uttorkning. Frånvaron av kontrollgrupper begränsar bedömningar av kausalitet, men det bör antas att perinatalt exponerade spädbarn kan löpa risk för biverkningar jämförbara med dem som rapporterats hos pediatriska och vuxna HIV-1-infekterade patienter som behandlats med lamivudin-innehållande kombinationsregimer. Långtidseffekter av exponering för lamivudin in utero och spädbarn är inte kända.
Erfarenhet efter marknadsföring
Följande biverkningar har identifierats efter användning av EPIVIR efter godkännande. Eftersom dessa reaktioner rapporteras frivilligt från en population av okänd storlek är det inte alltid möjligt att på ett tillförlitligt sätt uppskatta deras frekvens eller fastställa ett orsakssamband till läkemedelsexponering. Dessa reaktioner har valts för inkludering på grund av en kombination av deras allvarlighetsgrad, frekvens av rapportering eller potentiellt orsakssamband med lamivudin.
Kroppen som helhet
Omfördelning/ackumulering av kroppsfett.
Endokrina och metaboliska
Hyperglykemi.
Allmän
Svaghet. Hemisk och lymfatisk anemi (inklusive aplasi av ren röda blodkroppar och svår anemi som fortskrider under behandlingen).
Lever och bukspottkörtel
Laktacidos och leversteatos [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ], exacerbationer av hepatit B efter behandlingen [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Överkänslighet
Anafylaxi, urtikaria.
Muskuloskeletala
Muskelsvaghet, CPK-höjning, rabdomyolys. Hud Alopeci, klåda.
LÄKEMEDELSINTERAKTIONER
Läkemedel som hämmar organiska katjontransportörer
Lamivudin elimineras huvudsakligen i urinen genom aktiv organisk katjonisk sekretion. Möjligheten av interaktioner med andra läkemedel som administreras samtidigt bör övervägas, särskilt när deras huvudsakliga elimineringsväg är aktiv njursekretion via det organiska katjoniska transportsystemet (t.ex. trimetoprim) [se KLINISK FARMAKOLOGI ]. Det finns inga tillgängliga data om interaktioner med andra läkemedel som har njurclearance-mekanismer som liknar den för lamivudin.
Sorbitol
Samtidig administrering av engångsdoser av lamivudin och sorbitol resulterade i en dosberoende minskning av lamivudinexponeringen för sorbitol. Om möjligt, undvik användning av läkemedel som innehåller sorbitol tillsammans med lamivudin [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , KLINISK FARMAKOLOGI ].
VARNINGAR
Ingår som en del av FÖRSIKTIGHETSÅTGÄRDER sektion.
FÖRSIKTIGHETSÅTGÄRDER
Patienter med samtidig infektion med hepatit B-virus
Exacerbationer av hepatit efter behandling
Kliniska och laboratoriemässiga bevis för exacerbationer av hepatit har förekommit efter utsättning av lamivudin. Dessa exacerbationer har detekterats primärt av ALT-förhöjningar i serum utöver återuppkomst av HBV-DNA. Även om de flesta händelser verkar ha varit självbegränsande, har dödsfall rapporterats i vissa fall. Liknande händelser har rapporterats efter marknadsföring efter förändringar från lamivudin-innehållande HIV-1-behandlingsregimer till icke-lamivudin-innehållande regimer hos patienter infekterade med både HIV-1 och HBV. Orsakssambandet till utsättande av lamivudinbehandling är okänt. Patienter bör övervakas noggrant med både klinisk uppföljning och laboratorieuppföljning i minst flera månader efter avslutad behandling.
Viktiga skillnader mellan produkter som innehåller lamivudin
EPIVIR 150 mg tabletter och oral lösning innehåller en högre dos av samma aktiva ingrediens (lamivudin) än EPIVIR-HBV tabletter och EPIVIR-HBV oral lösning. EPIVIR-HBV utvecklades för patienter med kronisk hepatit B. Beredningen och doseringen av lamivudin i EPIVIR-HBV är inte lämpliga för patienter som samtidigt är infekterade med HIV-1 och HBV. Säkerhet och effekt av lamivudin har inte fastställts för behandling av kronisk hepatit B hos patienter som samtidigt är infekterade med HIV-1 och HBV. Om behandling med EPIVIR-HBV ordineras för kronisk hepatit B för en patient med okänd eller obehandlad HIV-1-infektion, kommer sannolikt snabb uppkomst av HIV-1-resistens att resultera på grund av den subterapeutiska dosen och olämpligheten av monoterapi HIV-1-behandling. Om ett beslut fattas att administrera lamivudin till patienter som samtidigt är infekterade med HIV-1 och HBV, ska EPIVIR 150 mg tabletter, EPIVIR 150 mg oral lösning eller annan produkt som innehåller den högre dosen lamivudin användas som en del av en lämplig kombinationsbehandling.
Uppkomst av Lamivudin-resistent HBV
Säkerhet och effekt av lamivudin har inte fastställts för behandling av kronisk hepatit B hos patienter dubbelt infekterade med HIV-1 och HBV (se fullständig förskrivningsinformation för EPIVIR-HBV). Uppkomsten av hepatit B-virusvarianter associerade med resistens mot lamivudin har också rapporterats hos HIV-1-infekterade försökspersoner som har fått lamivudin-innehållande antiretrovirala kurer i närvaro av samtidig infektion med hepatit B-virus.
Mjölkacidos och svår hepatomegali med Steatos
Laktacidos och svår hepatomegali med steatos, inklusive dödsfall, har rapporterats vid användning av nukleosidanaloger, inklusive EPIVIR. En majoritet av dessa fall har varit hos kvinnor. Kvinnligt kön och fetma kan vara riskfaktorer för utveckling av laktacidos och svår hepatomegali med steatos hos patienter som behandlas med antiretrovirala nukleosidanaloger. Behandling med EPIVIR bör avbrytas hos alla patienter som utvecklar kliniska fynd eller laboratoriefynd som tyder på laktacidos eller uttalad levertoxicitet, vilket kan inkludera hepatomegali och steatos även i frånvaro av markanta transaminashöjningar.
Använd med interferon- och ribavirinbaserade regimer
In vitro-studier har visat att ribavirin kan minska fosforyleringen av pyrimidinnukleosidanaloger såsom lamivudin. Även om inga tecken på en farmakokinetisk eller farmakodynamisk interaktion (t.ex. förlust av HIV-1/HCV-virologisk suppression) sågs när ribavirin administrerades samtidigt med lamivudin hos HIV-1/HCV-infekterade patienter [se KLINISK FARMAKOLOGI leverdekompensation (viss dödlig) har inträffat hos HIV-1/HCV-saminfekterade patienter som fått antiretroviral kombinationsbehandling för HIV-1 och interferon alfa med eller utan ribavirin. Patienter som får interferon alfa med eller utan ribavirin och EPIVIR bör övervakas noggrant med avseende på behandlingsrelaterade toxiciteter, särskilt leverdekompensation. Utsättning av EPIVIR 150 mg bör anses vara medicinskt lämpligt. Dosreduktion eller utsättande av interferon alfa, ribavirin eller båda bör också övervägas om förvärrade kliniska toxiciteter observeras, inklusive leverdekompensation (t.ex. Child-Pugh större än 6). Se hela förskrivningsinformationen för interferon och ribavirin.
Pankreatit
Hos pediatriska patienter med tidigare exponering för antiretroviral nukleosid, en historia av pankreatit eller andra betydande riskfaktorer för utveckling av pankreatit, bör EPIVIR 150 mg användas med försiktighet. Behandling med EPIVIR 150 mg ska avbrytas omedelbart om kliniska tecken, symtom eller laboratorieavvikelser som tyder på pankreatit uppstår [se NEGATIVA REAKTIONER ].
Immunrekonstitutionssyndrom
Immunrekonstitutionssyndrom har rapporterats hos patienter som behandlats med antiretroviral kombinationsterapi, inklusive EPIVIR. Under den inledande fasen av antiretroviral kombinationsbehandling kan patienter vars immunsystem svarar utveckla ett inflammatoriskt svar på indolenta eller kvarvarande opportunistiska infektioner (som Mycobacterium avium-infektion, cytomegalovirus, Pneumocystis jirovecii pneumoni [PCP] eller tuberkulos), vilket kan kräva ytterligare utvärdering och behandling.
Autoimmuna störningar (såsom Graves sjukdom, polymyosit och Guillain-Barrés syndrom) har också rapporterats förekomma i samband med immunrekonstitution, men tiden till debut är mer varierande och kan inträffa många månader efter påbörjad behandling.
Lägre virologisk suppressionsfrekvens och ökad risk för virusresistens med oral lösning
Pediatriska försökspersoner som fick EPIVIR 150 mg oral lösning (vid viktbandsbaserade doser på ungefär 8 mg per kg per dag) samtidigt med andra antiretrovirala orala lösningar vid någon tidpunkt i ARROW-studien hade lägre frekvenser av virologisk suppression, lägre plasmaexponering för lamivudin och utvecklade virusresistens oftare än de som får EPIVIR-tabletter [se KLINISK FARMAKOLOGI , Mikrobiologi , Kliniska studier ].
EPIVIR skårad tablett är den föredragna formuleringen för HIV-1-infekterade pediatriska patienter som väger minst 14 kg och för vilka en fast doseringsform är lämplig. En tablettbehandling bör användas när det är möjligt för att undvika en potentiell interaktion med sorbitol [se KLINISK FARMAKOLOGI ]. Överväg oftare övervakning av HIV-1 virusmängd vid behandling med EPIVIR 150 mg oral lösning.
Information om patientrådgivning
Rekommendera patienten att läsa den FDA-godkända patientmärkningen (patientinformation).
Patienter med hepatit B eller C samtidig infektion
Informera patienter som samtidigt är infekterade med HIV-1 och HBV att försämring av leversjukdom har inträffat i vissa fall när behandlingen med lamivudin avbröts. Råda patienterna att diskutera eventuella förändringar i behandlingen med sin vårdgivare [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Informera patienter med HIV-1/HCV samtidig infektion om att leverdekompensation (viss dödlig) har inträffat hos HIV-1/HCV co-infekterade patienter som får antiretroviral kombinationsbehandling för HIV-1 och interferon alfa med eller utan ribavirin [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Skillnader i formuleringar av EPIVIR
Informera patienterna om att EPIVIR 150 mg tabletter och oral lösning innehåller en högre dos av samma aktiva ingrediens (lamivudin) som EPIVIR-HBV tabletter och oral lösning. Om ett beslut fattas att inkludera lamivudin i HIV-1-behandlingsregimen för en patient som samtidigt är infekterad med HIV-1 och HBV, bör formuleringen och doseringen av lamivudin i EPIVIR (inte EPIVIR-HBV) användas [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Mjölkacidos/Hepatomegali med Steatos
Informera patienter om att laktacidos och svår hepatomegali med steatos har rapporterats vid användning av nukleosidanaloger och andra antiretrovirala medel. Råda patienter att sluta ta EPIVIR om de utvecklar kliniska symtom som tyder på laktacidos eller uttalad levertoxicitet [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Risk för pankreatit
Råda föräldrar eller vårdnadshavare att övervaka pediatriska patienter för tecken och symtom på pankreatit [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Immunrekonstitutionssyndrom
Rekommendera patienter att omedelbart informera sin vårdgivare om alla tecken och symtom på infektion eftersom inflammation från tidigare infektion kan uppstå strax efter antiretroviral kombinationsbehandling, inklusive när EPIVIR påbörjas [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Lägre virologisk suppressionsfrekvens och ökad risk för virusresistens med oral lösning
Informera patienterna om att en tablettbehandling bör användas när det är möjligt på grund av en ökad frekvens av behandlingsmisslyckande bland pediatriska patienter som fick EPIVIR oral lösning samtidigt med andra antiretrovirala orala lösningar [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Sackarosinnehåll i EPIVIR oral lösning
Informera diabetiker om att varje 15 ml dos av EPIVIR oral lösning innehåller 3 gram sackaros (1 ml = 200 mg sackaros) [se BESKRIVNING ].
Graviditetsregistret
Informera patienter om att det finns ett graviditetsexponeringsregister som övervakar graviditetsresultat hos kvinnor som exponerats för EPIVIR under graviditeten [se Användning i specifika populationer ].
Laktation
Instruera kvinnor med HIV-1-infektion att inte amma eftersom HIV-1 kan överföras till barnet i bröstmjölken [se Användning i specifika populationer ].
Missad dosering
Instruera patienterna att om de missar en dos av EPIVIR 150 mg, att ta den så snart de kommer ihåg det. Rekommendera patienter att inte dubbla sin nästa dos eller ta mer än den ordinerade dosen [se DOSERING OCH ADMINISTRERING ].
EPIVIR och RETROVIR är varumärken som ägs av eller licensieras till företagsgruppen ViiV Healthcare.
Icke-klinisk toxikologi
Karcinogenes, Mutagenes, Nedsatt fertilitet
Carcinogenes
Långtidsstudier av karcinogenicitet med lamivudin på möss och råttor visade inga tecken på karcinogen potential vid exponeringar upp till 10 gånger (möss) och 58 gånger (råttor) den humana exponeringen vid den rekommenderade dosen på 300 mg.
Mutagenes
Lamivudin var mutagent i en L5178Y muslymfomanalys och klastogen i en cytogenetisk analys med odlade humana lymfocyter. Lamivudin var inte mutagent i en mikrobiell mutagenicitetsanalys, i en celltransformationsanalys in vitro, i ett mikronukleustest på råtta, i en cytogenetisk analys av benmärg hos råtta och i en analys för oplanerad DNA-syntes i råttlever. Lamivudin visade inga tecken på genotoxisk aktivitet in vivo hos råtta vid orala doser på upp till 2 000 mg per kg, vilket gav plasmanivåer på 35 till 45 gånger högre än hos människa vid den rekommenderade dosen för HIV-1-infektion.
Nedsättning av fertilitet
I en studie av reproduktionsförmåga visade lamivudin administrerat till råttor i doser upp till 4 000 mg per kg per dag, vilket gav plasmanivåer 47 till 70 gånger högre än hos människor, inga tecken på nedsatt fertilitet och ingen effekt på överlevnad, tillväxt och utveckling till avvänjning av avkomman.
Användning i specifika populationer
Graviditet
Registret för graviditetsexponering
Det finns ett graviditetsexponeringsregister som övervakar graviditetsutfall hos kvinnor som exponerats för EPIVIR under graviditeten. Sjukvårdsleverantörer uppmuntras att registrera patienter genom att ringa Antiretroviral Pregnancy Registry (APR) på 1-800-258-4263.
Risksammanfattning
Tillgängliga data från APR visar ingen skillnad i den totala risken för fosterskador för lamivudin jämfört med bakgrundsfrekvensen för fosterskador på 2,7 % i referenspopulationen för Metropolitan Atlanta Congenital Defects Program (MACDP) (se Data ). APR använder MACDP som USA:s referenspopulation för fosterskador i den allmänna befolkningen. MACDP utvärderar kvinnor och spädbarn från ett begränsat geografiskt område och inkluderar inte resultat för förlossningar som inträffade vid mindre än 20 veckors graviditet. Frekvensen av missfall rapporteras inte i APR. Den uppskattade bakgrundsfrekvensen för missfall i kliniskt erkända graviditeter i den allmänna befolkningen i USA är 15 % till 20 %. Bakgrundsrisken för stora fosterskador och missfall för den indikerade populationen är okänd.
I reproduktionsstudier på djur resulterade oral administrering av lamivudin till dräktiga kaniner under organogenesen i embryodödlighet vid systemisk exponering (AUC) liknande den rekommenderade kliniska dosen; inga negativa utvecklingseffekter observerades dock vid oral administrering av lamivudin till dräktiga råttor under organogenesen vid plasmakoncentrationer (Cmax) 35 gånger den rekommenderade kliniska dosen (se Data ).
Data
Mänskliga data
Baserat på prospektiva rapporter till APR av över 11 000 exponeringar för lamivudin under graviditet som resulterade i levande födda barn (inklusive över 4 500 exponerade under första trimestern), fanns det ingen skillnad mellan den totala risken för fosterskador för lamivudin jämfört med frekvensen av fosterskador i bakgrunden av 2,7 % i den amerikanska referenspopulationen för MACDP. Prevalensen av defekter i levande födslar var 3,1 % (95 % KI: 2,6 % till 3,6 %) efter exponering för första trimestern för behandlingar innehållande lamivudin och 2,8 % (95 % KI: 2,5 % till 3,3 %) efter exponering för andra/tredje trimestern till regimer som innehåller lamivudin.
Lamivudins farmakokinetik studerades hos gravida kvinnor under 2 kliniska prövningar utförda i Sydafrika. Studierna utvärderade farmakokinetiken hos 16 kvinnor vid 36 veckors graviditet som använde 150 mg lamivudin två gånger dagligen med zidovudin, 10 kvinnor vid 38 veckors graviditet som använde 150 mg lamivudin två gånger dagligen med zidovudin och 10 kvinnor vid 38 veckors graviditet två gånger dagligen med 300 mg lamivudin. dagligen utan andra antiretrovirala medel. Dessa försök var inte utformade eller drivna för att ge information om effektivitet. Lamivudinkoncentrationerna var i allmänhet likartade i serumprover från modern, neonatal och navelsträng. Hos en undergrupp av försökspersoner togs fostervattenprover efter naturligt membranbrott och bekräftade att lamivudin passerar placentan hos människor. Baserat på begränsade data vid förlossningen, var median (intervall) fostervattenkoncentrationer av lamivudin 3,9 (1,2 till 12,8) gånger högre jämfört med parad maternell serumkoncentration (n = 8).
Djurdata
Lamivudin administrerades oralt till gravida råttor (vid 90, 600 och 4 000 mg per kg per dag) och kaniner (vid 90, 300 och 1 000 mg per kg per dag och vid 15, 40 och 90 mg per kg per dag) under organogenes (på dräktighet dag 7 till 16 [råtta] och 8 till 20 [kanin]). Inga tecken på fostermissbildningar på grund av lamivudin observerades hos råttor och kaniner vid doser som gav plasmakoncentrationer (Cmax) cirka 35 gånger högre än exponering för människa vid den rekommenderade dagliga dosen. Bevis på tidig embryodödlighet sågs hos kanin vid systemexponeringar (AUC) liknande de som observerats hos människor, men det fanns ingen indikation på denna effekt hos råtta vid plasmakoncentrationer (Cmax) 35 gånger högre än mänsklig exponering vid den rekommenderade dagliga dosen . Studier på gravida råttor visade att lamivudin överförs till fostret genom moderkakan. I fertilitets-/för- och postnatal utvecklingsstudie på råttor, administrerades lamivudin oralt i doser på 180, 900 och 4 000 mg per kg och dag (från före parning till postnatal dag 20). I studien påverkades inte avkommans utveckling, inklusive fertilitet och reproduktionsförmåga, av moderns administrering av lamivudin.
Laktation
Risksammanfattning
Centers for Disease Control and Prevention rekommenderar att HIV-1-infekterade mödrar i USA inte ammar sina spädbarn för att undvika att riskera postnatal överföring av HIV-1-infektion. Lamivudin finns i modersmjölk. Det finns ingen information om effekterna av lamivudin på det ammade barnet eller läkemedlens effekter på mjölkproduktionen. På grund av risken för (1) HIV-1-överföring (hos HIV-negativa spädbarn), (2) utveckla virusresistens (hos HIV-positiva spädbarn) och (3) biverkningar hos ett ammat spädbarn, instruera mammor att inte amma om de får EPIVIR.
Pediatrisk användning
Säkerheten och effektiviteten av EPIVIR 150 mg i kombination med andra antiretrovirala medel har fastställts hos pediatriska patienter i åldern 3 månader och äldre. EPIVIR skårad tablett är den föredragna formuleringen för HIV-1-infekterade pediatriska patienter som väger minst 14 kg och för vilka en fast doseringsform är lämplig eftersom pediatriska försökspersoner som fick EPIVIR 150 mg oral lösning hade lägre frekvens av virologisk suppression, lägre plasmaexponering för lamivudin , och utvecklade virusresistens oftare än de som fick EPIVIR 150 mg tabletter i ARROW-studien [se DOSERING OCH ADMINISTRERING , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , NEGATIVA REAKTIONER , KLINISK FARMAKOLOGI , Kliniska studier ].
Geriatrisk användning
Kliniska prövningar av EPIVIR inkluderade inte tillräckligt många försökspersoner i åldern 65 år och äldre för att avgöra om de svarar annorlunda än yngre försökspersoner. Generellt sett bör försiktighet iakttas vid administrering av EPIVIR till äldre patienter, vilket återspeglar den högre frekvensen av nedsatt lever-, njur- eller hjärtfunktion och av samtidig sjukdom eller annan läkemedelsbehandling [se DOSERING OCH ADMINISTRERING , KLINISK FARMAKOLOGI ].
Patienter med nedsatt njurfunktion
Reduktion av dosen av EPIVIR 150 mg rekommenderas för patienter med nedsatt njurfunktion [se DOSERING OCH ADMINISTRERING , KLINISK FARMAKOLOGI ].
ÖVERDOS
Det finns ingen känd specifik behandling för överdosering med EPIVIR. Om överdosering inträffar ska patienten övervakas och standardstödjande behandling tillämpas vid behov. Eftersom en försumbar mängd lamivudin avlägsnades via (4-timmars) hemodialys, kontinuerlig ambulatorisk peritonealdialys och automatiserad peritonealdialys, är det inte känt om kontinuerlig hemodialys skulle ge klinisk nytta vid en överdos av lamivudin.
KONTRAINDIKATIONER
EPIVIR 150 mg är kontraindicerat hos patienter med en tidigare överkänslighetsreaktion mot lamivudin.
KLINISK FARMAKOLOGI
Handlingsmekanism
Lamivudin är ett antiretroviralt medel [se Mikrobiologi ].
Farmakokinetik
Farmakokinetik hos vuxna
De farmakokinetiska egenskaperna hos lamivudin har studerats hos asymtomatiska, HIV-1-infekterade vuxna försökspersoner efter administrering av enstaka intravenösa (IV) doser från 0,25 till 8 mg per kg, såväl som enstaka och multipla (två gånger dagligen) orala doser från 0,25 till 10 mg per kg.
De farmakokinetiska egenskaperna hos lamivudin har också studerats som enstaka och multipla orala doser från 5 mg till 600 mg per dag administrerade till HBV-infekterade patienter.
De farmakokinetiska egenskaperna vid steady-state hos EPIVIR 300 mg tabletten en gång dagligen i 7 dagar jämfört med EPIVIR 150 mg tabletten två gånger dagligen i 7 dagar utvärderades i en crossover-studie med 60 friska försökspersoner. EPIVIR 300 mg en gång dagligen resulterade i lamivudinexponeringar som liknade EPIVIR 150 mg två gånger dagligen med avseende på plasma AUC24,ss; Cmax,ss var dock 66 % högre och dalvärdet 53 % lägre jämfört med 150 mg två gånger dagligen. Intracellulära lamivudintrifosfatexponeringar i perifera mononukleära blodceller var också likartade med avseende på AUC24,ss och Cmax24,ss; dock var dalvärden lägre jämfört med 150 mg två gånger dagligen. Variabiliteten mellan patienter var större för intracellulära lamivudintrifosfatkoncentrationer jämfört med dalkoncentrationer av lamivudin i plasma.
Farmakokinetiken för lamivudin utvärderades hos 12 vuxna HIV-1-infekterade försökspersoner som doserades med lamivudin 150 mg två gånger dagligen i kombination med andra antiretrovirala medel. Det geometriska medelvärdet (95 % KI) för AUC(0-12) var 5,53 (4,58, 6,67) mcg.h per ml och för Cmax var 1,40 (1,17, 1,69) mcg per ml.
Absorption och biotillgänglighet
Absolut biotillgänglighet hos 12 vuxna försökspersoner var 86 % ± 16 % (medelvärde ± SD) för 150 mg tabletten och 87 % ± 13 % för oral lösning. Efter oral administrering av 2 mg per kg två gånger dagligen till 9 vuxna med HIV-1 var den maximala serumkoncentrationen av lamivudin (Cmax) 1,5 ± 0,5 mcg per ml (medelvärde ± SD). Ytan under kurvan för plasmakoncentration mot tid (AUC) och Cmax ökade i proportion till oral dos inom intervallet från 0,25 till 10 mg per kg.
Ackumuleringskvoten för lamivudin hos HIV-1-positiva asymtomatiska vuxna med normal njurfunktion var 1,50 efter 15 dagars oral administrering av 2 mg per kg två gånger dagligen.
Effekter av mat på oral absorption
EPIVIR 150 mg tabletter och oral lösning kan administreras med eller utan mat. En undersökningsdosform på 25 mg av lamivudin administrerades oralt till 12 asymtomatiska, HIV-1-infekterade försökspersoner vid 2 tillfällen, en gång i fastande tillstånd och en gång med mat (1 099 kcal; 75 gram fett, 34 gram protein, 72 gram kolhydrater ). Absorptionen av lamivudin var långsammare i utfodrat tillstånd (Tmax: 3,2 ± 1,3 timmar) jämfört med fastande (Tmax: 0,9 ± 0,3 timmar); Cmax i matat tillstånd var 40 % ± 23 % (medelvärde ± SD) lägre än i fastande tillstånd. Det fanns ingen signifikant skillnad i systemisk exponering (AUC∞) i föda och fastande tillstånd.
Distribution
Den skenbara distributionsvolymen efter intravenös administrering av lamivudin till 20 försökspersoner var 1,3 ± 0,4 l per kg, vilket tyder på att lamivudin distribueras till extravaskulära utrymmen. Distributionsvolymen var oberoende av dos och korrelerade inte med kroppsvikten.
Bindningen av lamivudin till humana plasmaproteiner är mindre än 36 %. In vitro-studier visade att över koncentrationsintervallet 0,1 till 100 mcg per ml varierade mängden lamivudin associerad med erytrocyter från 53 % till 57 % och var oberoende av koncentrationen.
Ämnesomsättning
Metabolism av lamivudin är en mindre elimineringsväg. Hos människor är den enda kända metaboliten av lamivudin transsulfoxidmetaboliten (cirka 5 % av en oral dos efter 12 timmar). Serumkoncentrationer av denna metabolit har inte fastställts. Lamivudin metaboliseras inte signifikant av cytokrom P450-enzymer.
Eliminering
Majoriteten av lamivudin elimineras oförändrat i urinen genom aktiv organisk katjonisk sekretion. Hos 9 friska försökspersoner som fick en engångsdos på 300 mg peroral lamivudin var renalt clearance 199,7 ± 56,9 ml per min (medelvärde ± SD). Hos 20 HIV-1-infekterade försökspersoner som fick en enda IV-dos var njurclearance 280,4 ± 75,2 ml per min (medelvärde ± SD), vilket motsvarar 71 % ± 16 % (medelvärde ± SD) av totalt clearance av lamivudin.
I de flesta endosstudier på HIV-1-infekterade försökspersoner, HBV-infekterade försökspersoner eller friska försökspersoner med serumprovtagning under 24 timmar efter dosering, varierade den observerade genomsnittliga eliminationshalveringstiden (t½) från 5 till 7 timmar. Hos HIV-1-infekterade försökspersoner var totalt clearance 398,5 ± 69,1 ml per minut (medelvärde ± SD). Oralt clearance och eliminationshalveringstid var oberoende av dos och kroppsvikt över ett oralt doseringsintervall på 0,25 till 10 mg per kg.
Specifika populationer
Patienter med nedsatt njurfunktion
De farmakokinetiska egenskaperna för lamivudin har fastställts hos en liten grupp HIV-1-infekterade vuxna med nedsatt njurfunktion (tabell 7).
Tmax påverkades inte signifikant av njurfunktionen. Baserat på dessa observationer rekommenderas att dosen av lamivudin ändras hos patienter med nedsatt njurfunktion [se DOSERING OCH ADMINISTRERING ].
Baserat på en studie på i övrigt friska försökspersoner med nedsatt njurfunktion, ökade hemodialys lamivudinclearance från i genomsnitt 64 till 88 ml per minut; dock var hemodialystiden (4 timmar) otillräcklig för att signifikant ändra den genomsnittliga lamivudinexponeringen efter en engångsdos. Kontinuerlig ambulatorisk peritonealdialys och automatiserad peritonealdialys har försumbar effekt på lamivudinclearance. Därför rekommenderas det, efter korrigering av dosen för kreatininclearance, att ingen ytterligare dosändring görs efter rutinmässig hemodialys eller peritonealdialys.
Effekterna av nedsatt njurfunktion på lamivudins farmakokinetik hos pediatriska patienter är inte kända.
Patienter med nedsatt leverfunktion
De farmakokinetiska egenskaperna för lamivudin har fastställts hos vuxna med nedsatt leverfunktion. Farmakokinetiska parametrar förändrades inte av försämrad leverfunktion. Säkerhet och effekt av lamivudin har inte fastställts i närvaro av dekompenserad leversjukdom.
Gravid kvinna
Lamivudins farmakokinetik studerades hos 36 gravida kvinnor under 2 kliniska prövningar utförda i Sydafrika. Lamivudins farmakokinetik hos gravida kvinnor liknade den som ses hos icke-gravida vuxna och hos kvinnor efter förlossningen. Lamivudinkoncentrationerna var i allmänhet likartade i serumprover från modern, neonatal och navelsträng.
Pediatriska patienter
Farmakokinetiken för lamivudin har studerats efter antingen enstaka eller upprepade doser av EPIVIR hos 210 pediatriska försökspersoner. Pediatriska försökspersoner som fick lamivudin oral lösning (doserad med cirka 8 mg per kg per dag) uppnådde cirka 25 % lägre plasmakoncentrationer av lamivudin jämfört med HIV-1-infekterade vuxna. Pediatriska försökspersoner som fick lamivudin orala tabletter uppnådde plasmakoncentrationer jämförbara med eller något högre än de som observerats hos vuxna. Den absoluta biotillgängligheten för både EPIVIR tabletter och oral lösning är lägre hos barn än hos vuxna. Den relativa biotillgängligheten för EPIVIR oral lösning är cirka 40 % lägre än tabletter som innehåller lamivudin hos pediatriska försökspersoner, trots ingen skillnad hos vuxna. Lägre lamivudinexponeringar hos pediatriska patienter som får EPIVIR 150 mg oral lösning beror sannolikt på interaktionen mellan lamivudin och samtidiga lösningar som innehåller sorbitol (såsom ZIAGEN). Modellering av farmakokinetiska data tyder på att det behövs en ökning av dosen av EPIVIR oral lösning till 5 mg per kg oralt två gånger dagligen eller 10 mg per kg oralt en gång dagligen (upp till maximalt 300 mg dagligen) för att uppnå tillräckliga koncentrationer av lamivudin [se DOSERING OCH ADMINISTRERING ]. Det finns inga kliniska data från HIV-1-infekterade pediatriska patienter som administreras samtidigt med läkemedel som innehåller sorbitol i denna dos.
Farmakokinetiken för lamivudin doserat en gång dagligen till HIV-1-infekterade pediatriska försökspersoner i åldern 3 månader till 12 år utvärderades i 3 studier (PENTA-15 [n = 17], PENTA 13 [n = 19] och ARROW PK [n = 35]). Alla 3 studierna var 2-perioder, crossover, öppna farmakokinetiska prövningar med två gånger kontra en gång dagligen dosering av abakavir och lamivudin. Dessa 3 studier visade att dosering en gång dagligen ger liknande AUC0-24 till dosering två gånger dagligen av lamivudin vid samma totala dagliga dos när man jämförde doseringsregimerna inom samma formulering (dvs. antingen oral lösning eller tablettformulering). Medelvärdet för Cmax var cirka 80 % till 90 % högre med lamivudin en gång dagligen jämfört med dosering två gånger dagligen.
Distributionen av lamivudin i cerebrospinalvätska (CSF) utvärderades hos 38 pediatriska försökspersoner efter upprepad oral dosering med lamivudin. CSF-prover samlades in mellan 2 och 4 timmar efter dosering. Vid dosen 8 mg per kg per dag varierade CSF-lamivudinkoncentrationerna hos 8 försökspersoner från 5,6 % till 30,9 % (medelvärde ± SD på 14,2 % ± 7,9 %) av koncentrationen i ett samtidigt serumprov, med CSF-lamivudinkoncentrationer från 0,04 till 0,3 mcg per ml.
Begränsade, okontrollerade farmakokinetiska data och säkerhetsdata finns tillgängliga från administrering av lamivudin (och zidovudin) till 36 spädbarn i åldern upp till 1 vecka i 2 studier i Sydafrika. I dessa studier reducerades lamivudinclearance avsevärt hos 1 vecka gamla nyfödda jämfört med pediatriska försökspersoner (över 3 månader) som studerats tidigare. Det finns otillräcklig information för att fastställa tidsförloppet för förändringar i clearance mellan den omedelbara neonatalperioden och åldersintervallen över 3 månader gamla [se NEGATIVA REAKTIONER ].
Geriatriska patienter
Farmakokinetiken för lamivudin efter administrering av EPIVIR 150 mg till personer över 65 år har inte studerats [se Användning i specifika populationer ].
Manliga Och Kvinnliga Patienter
Det finns inga signifikanta eller kliniskt relevanta könsskillnader i lamivudins farmakokinetik.
Rasgrupper
Det finns inga signifikanta eller kliniskt relevanta rasskillnader i lamivudins farmakokinetik.
Läkemedelsinteraktionsstudier
Effekt av Lamivudin på farmakokinetiken hos andra medel
Baserat på in vitro-studieresultat förväntas inte lamivudin vid terapeutisk läkemedelsexponering påverka farmakokinetiken för läkemedel som är substrat för följande transportörer: organisk anjontransportörpolypeptid 1B1/3 (OATP1B1/3), bröstcancerresistensprotein (BCRP), P-glykoprotein (P-gp), multidrog- och toxinextruderingsprotein 1 (MATE)1, MATE2-K, organisk katjontransportör 1 (OCT)1, OCT2 eller OCT3.
Effekt av andra medel på farmakokinetiken för lamivudin
Lamivudin är ett substrat för MATE1, MATE2-K och OCT2 in vitro. Trimetoprim (en hämmare av dessa läkemedelstransportörer) har visats öka plasmakoncentrationerna av lamivudin. Denna interaktion anses inte vara kliniskt signifikant eftersom ingen dosjustering av lamivudin behövs.
Lamivudin är ett substrat för P-gp och BCRP; med tanke på dess absoluta biotillgänglighet (87 %) är det dock osannolikt att dessa transportörer spelar en signifikant roll vid absorptionen av lamivudin. Därför är det osannolikt att samtidig administrering av läkemedel som hämmar dessa effluxtransportörer påverkar dispositionen och elimineringen av lamivudin.
Interferon Alfa
Det fanns ingen signifikant farmakokinetisk interaktion mellan lamivudin och interferon alfa i en studie med 19 friska manliga försökspersoner [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Ribavirin
In vitro-data indikerar att ribavirin minskar fosforyleringen av lamivudin, stavudin och zidovudin. Ingen farmakokinetisk (t.ex. plasmakoncentrationer eller intracellulära trifosforylerade aktiva metaboliter) eller farmakodynamiska (t.ex. förlust av HIV-1/HCV virologisk suppression) interaktion observerades när ribavirin och lamivudin (n = 18), stavudin (n = 10) , eller zidovudin (n = 6) administrerades samtidigt som en del av en multidrogregim till HIV-1/HCV-saminfekterade försökspersoner [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Sorbitol (hjälpämne)
Lamivudin- och sorbitollösningar administrerades samtidigt till 16 friska vuxna försökspersoner i en öppen, randomiserad sekvens, 4-periods, crossover-studie. Varje individ fick en enstaka 300 mg dos av lamivudin oral lösning ensam eller samtidigt administrerad med en engångsdos på 3,2 gram, 10,2 gram eller 13,4 gram sorbitol i lösning. Samtidig administrering av lamivudin och sorbitol resulterade i dosberoende minskningar med 20 %, 39 % och 44 % i AUC(0-24), 14 %, 32 % och 36 % i AUC(∞) och 28 %, 52 % och 55 % i Cmax; av lamivudin, respektive.
Trimetoprim/sulfametoxazol
Lamivudin och TMP/SMX administrerades samtidigt till 14 HIV-1-positiva försökspersoner i en enkelcenter, öppen, randomiserad, crossover-studie. Varje individ fick behandling med en engångsdos på 300 mg lamivudin och TMP 160 mg/SMX 800 mg en gång dagligen i 5 dagar med samtidig administrering av lamivudin 300 mg med den femte dosen i en crossover-design. Samtidig administrering av TMP/SMX med lamivudin resulterade i en ökning med 43 % ± 23 % (medelvärde ± SD) i lamivudin AUC∞, en minskning med 29 % ± 13 % i oral clearance av lamivudin och en minskning med 30 % ± 36 % i renal clearance av lamivudin. De farmakokinetiska egenskaperna för TMP och SMX förändrades inte vid samtidig administrering med lamivudin. Det finns ingen information om effekten på lamivudins farmakokinetik av högre doser av TMP/SMX, såsom de som används vid behandling av PCP.
Zidovudin
Inga kliniskt signifikanta förändringar i lamivudins eller zidovudins farmakokinetik observerades hos 12 asymtomatiska HIV-1-infekterade vuxna försökspersoner som fick en singeldos zidovudin (200 mg) i kombination med flera doser lamivudin (300 mg var 12:e timme).
Mikrobiologi
Handlingsmekanism
Lamivudin är en syntetisk nukleosidanalog. Intracellulärt fosforyleras lamivudin till dess aktiva 5'-trifosfatmetabolit, lamivudintrifosfat (3TC-TP). Det huvudsakliga verkningssättet för 3TC-TP är hämning av HIV-1 omvänt transkriptas (RT) via DNA-kedjeterminering efter inkorporering av nukleotidanalogen.
Antiviral aktivitet
Den antivirala aktiviteten av lamivudin mot HIV-1 utvärderades i ett antal cellinjer inklusive monocyter och färska humana perifera blodlymfocyter (PBMC) med hjälp av standardkänslighetsanalyser. EC50-värdena låg i intervallet 0,003 till 15 mikroM (1 mikroM = 0,23 mikrogram per ml). Medianvärdet för EC50 för lamivudin var 60 nM (intervall: 20 till 70 nM), 35 nM (intervall: 30 till 40 nM), 30 nM (intervall: 20 till 90 nM), 20 nM (intervall: 3 till 40 nM) 30 nM (intervall: 1 till 60 nM), 30 nM (intervall: 20 till 70 nM), 30 nM (intervall: 3 till 70 nM) och 30 nM (intervall: 20 till 90 nM) mot HIV-1-klader AG- och grupp O-virus (n = 3 förutom n = 2 för klass B). EC50-värdena mot HIV-2-isolat (n = 4) varierade från 0,003 till 0,120 mikroM i PBMC. Lamivudin var inte antagonistiskt mot alla testade anti-HIV-medel. Ribavirin (50 mikroM) som används vid behandling av kronisk HCV-infektion minskade anti-HIV-1-aktiviteten hos lamivudin med 3,5 gånger i MT-4-celler.
Motstånd
Lamivudinresistenta varianter av HIV-1 har selekterats i cellodling. Genotypisk analys visade att resistensen berodde på en specifik aminosyrasubstitution i HIV-1 omvänt transkriptas vid kodon 184 som ändrade metioninet till antingen valin eller isoleucin (M184V/I).
HIV-1-stammar som är resistenta mot både lamivudin och zidovudin har isolerats från försökspersoner. Kliniska isolats mottaglighet för lamivudin och zidovudin övervakades i kontrollerade kliniska prövningar. Hos patienter som fick lamivudin som monoterapi eller kombinationsbehandling med lamivudin plus zidovudin, blev HIV-1-isolat från de flesta försökspersoner fenotypiskt och genotypiskt resistenta mot lamivudin inom 12 veckor.
Genotypisk och fenotypisk analys av HIV-1-isolat under behandling från försökspersoner med virologiskt misslyckande
Prov EPV20001
Femtiotre av 554 (10 %) försökspersoner inskrivna i EPV20001 identifierades som virologiska misslyckanden (plasma HIV-1 RNA-nivå högre än eller lika med 400 kopior per ml) i vecka 48. Tjugoåtta försökspersoner randomiserades till lamivudin en gång- daglig behandlingsgrupp och 25 till lamivudinbehandlingsgruppen två gånger dagligen. Medianvärdet för plasmanivåerna av HIV-1 RNA hos patienter i gruppen lamivudin en gång dagligen och lamivudin två gånger dagligen var 4,9 log10 kopior per ml respektive 4,6 log10 kopior per ml.
Genotypisk analys av isolat under behandling från 22 försökspersoner som identifierats som virologiska misslyckanden i gruppen lamivudin en gång dagligen visade att isolat från 8 av 22 försökspersoner innehöll en behandlingsuppkommande lamivudinresistensassocierad substitution (M184V eller M184I), isolat från 0 av 22 försökspersoner innehöll behandlingsuppkommande aminosyrasubstitutioner associerade med zidovudinresistens (M41L, D67N, K70R, L210W, T215Y/F eller K219Q/E), och isolat från 10 av 22 försökspersoner innehöll behandlingsuppkomna aminosyrasubstitutioner associerade med efavirenzresistens ( L100I, K101E, K103N, V108I eller Y181C).
Genotypisk analys av isolat under behandling från försökspersoner (n = 22) i lamivudinbehandlingsgruppen två gånger dagligen visade att isolat från 5 av 22 försökspersoner innehöll behandlingsuppkomna lamivudinresistenssubstitutioner, isolat från 1 av 22 försökspersoner innehöll behandlingsuppkommande zidovudinresistens substitutioner och isolat från 7 av 22 försökspersoner innehöll behandlingsuppkomna efavirenzresistenssubstitutioner.
Fenotypisk analys av baseline-matchade HIV-1-isolat under behandling från försökspersoner (n = 13) som fick lamivudin en gång dagligen visade att isolat från 7 av 13 försökspersoner visade en 85- till 299-faldig minskning av känsligheten för lamivudin, isolat från 12 av 13 försökspersoner var mottagliga för zidovudin och isolat från 8 av 13 försökspersoner uppvisade en 25- till 295-faldig minskning av känsligheten för efavirenz.
Fenotypisk analys av baseline-matchade HIV-1-isolat under behandling från försökspersoner (n = 13) som fick lamivudin två gånger dagligen visade att isolat från 4 av 13 försökspersoner uppvisade en 29- till 159-faldig minskning av känsligheten för lamivudin, isolat från alla 13 försökspersonerna var mottagliga för zidovudin och isolat från 3 av 13 försökspersoner uppvisade en 21- till 342-faldig minskning av känsligheten för efavirenz.
Prova EPV40001
Femtio försökspersoner fick lamivudin 300 mg en gång dagligen plus zidovudin 300 mg två gånger dagligen plus abakavir 300 mg två gånger dagligen och 50 försökspersoner fick lamivudin 150 mg plus zidovudin 300 mg plus abakavir 300 mg två gånger dagligen. Medianhalten av HIV-1 RNA i plasma vid baslinjen för försökspersoner i de två grupperna var 4,79 log10 kopior per ml respektive 4,83 log10 kopior per ml. Fjorton av 50 försökspersoner i gruppen som behandlades med lamivudin en gång dagligen och 9 av 50 försökspersoner i gruppen med lamivudin två gånger dagligen identifierades som virologiska misslyckanden.
Genotypisk analys av HIV-1-isolat under behandling från försökspersoner (n = 9) i gruppen som behandlades med lamivudin en gång dagligen visade att isolat från 6 försökspersoner hade en abakavir- och/eller lamivudinresistensrelaterad substitution M184V enbart. Isolat under behandling från försökspersoner (n = 6) som fick lamivudin två gånger dagligen visade att isolat från 2 försökspersoner hade M184V enbart, och isolat från 2 försökspersoner hade M184V-substitutionen i kombination med zidovudinresistensassocierade aminosyrasubstitutioner.
Fenotypisk analys av isolat under behandling från försökspersoner (n = 6) som fick lamivudin en gång dagligen visade att HIV-1-isolat från 4 försökspersoner uppvisade en 32- till 53-faldig minskning av känsligheten för lamivudin. HIV-1-isolat från dessa 6 försökspersoner var mottagliga för zidovudin.
Fenotypisk analys av isolat under behandling från försökspersoner (n = 4) som fick lamivudin två gånger dagligen visade att HIV-1-isolat från 1 försöksperson uppvisade en 45-faldig minskning av känsligheten för lamivudin och en 4,5-faldig minskning av känsligheten för zidovudin.
Pediatrik
Pediatriska försökspersoner som fick lamivudin oral lösning samtidigt med andra antiretrovirala orala lösningar (abakavir, nevirapin/efavirenz eller zidovudin) i ARROW utvecklade virusresistens oftare än de som fick tabletter. Vid randomisering till dosering en gång dagligen eller två gånger dagligen av EPIVIR plus abakavir hade 13 % av försökspersonerna som började med tabletter och 32 % av försökspersonerna som började med lösning resistenssubstitutioner. Resistensprofilen som observerats inom pediatrik liknar den som observerats hos vuxna när det gäller de detekterade genotypsubstitutionerna och relativ frekvens, med de vanligaste detekterade substitutionerna vid M184 (V eller I) [se Kliniska studier ].
Korsmotstånd
Korsresistens har observerats bland nukleosid omvänt transkriptashämmare (NRTI). Lamivudinresistenta HIV-1-mutanter var korsresistenta i cellkultur mot didanosin (ddI). Korsresistens förväntas också med abakavir och emtricitabin eftersom dessa väljer M184V-substitutioner.
Kliniska studier
Användningen av EPIVIR baseras på resultaten från kliniska prövningar på HIV-1-infekterade försökspersoner i kombinationsregimer med andra antiretrovirala medel. Information från prövningar med kliniska effektmått eller en kombination av CD4+-cellantal och HIV-1 RNA-mätningar ingår nedan som dokumentation av lamivudins bidrag till en kombinationsbehandling i kontrollerade prövningar.
Vuxna ämnen
Clinical Endpoint Trial
NUCB3007 (CAESAR) var en multicenter, dubbelblind, placebokontrollerad studie som jämförde fortsatt pågående behandling (enbart zidovudin [62 % av försökspersonerna] eller zidovudin med didanosin eller zalcitabin [38 % av försökspersonerna]) med tillägg av EPIVIR 150 mg eller EPIVIR 150 mg plus en icke-nukleosid omvänt transkriptashämmare (NNRTI), randomiserad 1:2:1. Totalt 1 816 HIV-1-infekterade vuxna med 25 till 250 CD4+-celler per mm³ (median = 122 celler per mm³) vid baslinjen: medianåldern var 36 år, 87% var män, 84% var nukleosid-erfarna, och 16 % var terapinaiva. Mediantiden för försöket var 12 månader. Resultaten sammanfattas i tabell 9.
Surrogatslutpunktsförsök
Dubbla nukleosidanalogförsök
Huvudsakliga kliniska prövningar i den initiala utvecklingen av lamivudin jämförde lamivudin/zidovudinkombinationer med zidovudin monoterapi eller med zidovudin plus zalcitabin. Dessa studier visade den antivirala effekten av lamivudin i en kombination av två läkemedel. Senare användningar av lamivudin vid behandling av HIV-1-infektion inkluderar det i flera läkemedelsregimer som innehåller minst 3 antiretrovirala läkemedel för förbättrad virussuppression.
Jämförelse av dosregimer Surrogatändpunktsförsök hos terapinaiva vuxna
EPV20001 var en multicenter, dubbelblind, kontrollerad studie där försökspersoner randomiserades 1:1 för att få EPIVIR 300 mg en gång dagligen eller EPIVIR 150 mg två gånger dagligen, i kombination med zidovudin 300 mg två gånger dagligen och efavirenz 600 mg en gång dagligen. Totalt 554 antiretrovirala behandlingsnaiva HIV-1-infekterade vuxna inkluderade: män (79 %), vita (50 %), medianålder 35 år, CD4+-cellantal vid baslinjen på 69 till 1 089 celler per mm³ (median = 362 celler per mm³), och medianbaslinjeplasma HIV-1 RNA på 4,66 log10 kopior per ml. Resultaten av behandlingen under 48 veckor sammanfattas i figur 1 och tabell 10.
Figur 1: Virologisk respons till och med vecka 48, EPV20001a,b (Intent-to-Treat) 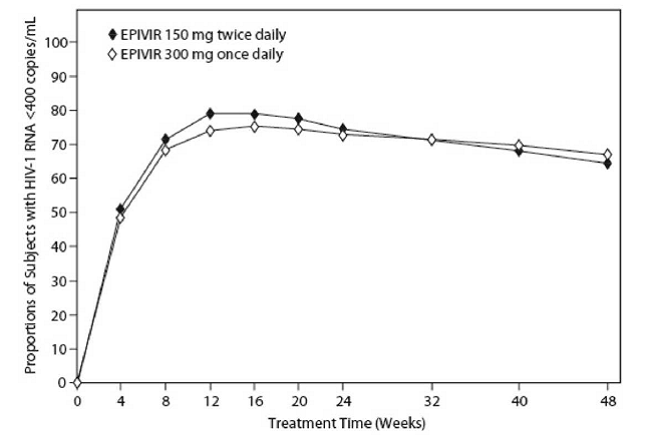
en Roche AMPLICOR HIV-1 MONITOR. b Responders vid varje besök är försökspersoner som hade uppnått och bibehållit HIV-1 RNA mindre än 400 kopior per ml utan att avbryta behandlingen vid det besöket.
Andelen försökspersoner med HIV-1 RNA mindre än 50 kopior per ml (via Roche Ultrasensitive analys) till och med vecka 48 var 61 % för försökspersoner som fick EPIVIR 300 mg en gång dagligen och 63 % för försökspersoner som fick EPIVIR 150 mg två gånger dagligen. Medianökningen av CD4+-cellantal var 144 celler per mm³ vid vecka 48 hos försökspersoner som fick EPIVIR 300 mg en gång dagligen och 146 celler per mm³ för försökspersoner som fick EPIVIR 150 mg två gånger dagligen.
En liten, randomiserad, öppen pilotstudie, EPV40001, genomfördes i Thailand. Totalt 159 behandlingsnaiva vuxna försökspersoner (män 32 %, asiatiska 100 %, medianålder 30 år, baseline median CD4+ cellantal 380 celler per mm³, median plasma HIV-1 RNA 4,8 log10 kopior per ml) registrerades. Två av behandlingsarmarna i denna studie gav en jämförelse mellan lamivudin 300 mg en gång dagligen (n = 54) och lamivudin 150 mg två gånger dagligen (n = 52), var och en i kombination med zidovudin 300 mg två gånger dagligen och abakavir 300 mg två gånger dagligen. I intent-to-treat-analyser av 48-veckorsdata var andelen försökspersoner med HIV-1 RNA under 400 kopior per ml 61 % (33 av 54) i gruppen som randomiserades till lamivudin en gång dagligen och 75 % (39 av 54) 52) i gruppen som randomiserades till att få alla 3 läkemedlen två gånger dagligen; proportionerna med HIV-1 RNA under 50 kopior per ml var 54 % (29 av 54) i gruppen med lamivudin en gång dagligen och 67 % (35 av 52) i gruppen med alla två gånger dagligen; och medianökningen av CD4+-cellantal var 166 celler per mm³ i gruppen med lamivudin en gång dagligen och 216 celler per mm³ i gruppen som fick alla två gånger dagligen.
Pediatriska ämnen
Clinical Endpoint Trial
ACTG300 var en multicenter, randomiserad, dubbelblind studie som gjorde det möjligt att jämföra EPIVIR plus RETROVIR (zidovudin) med didanosin som monoterapi. Totalt 471 symtomatiska, HIV-1-infekterade terapinaiva (mindre än eller lika med 56 dagars antiretroviral terapi) pediatriska patienter inkluderades i dessa två behandlingsgrupper. Medianåldern var 2,7 år (intervall: 6 veckor till 14 år), 58 % var kvinnor och 86 % var icke-vita. Det genomsnittliga antalet CD4+-celler vid baslinjen var 868 celler per mm³ (medelvärde: 1 060 celler per mm³ och intervall: 0 till 4 650 celler per mm³ för försökspersoner som är mindre än eller lika med 5 år; medelvärde: 419 celler per mm³ och intervall: 0 till 1 555 celler per mm³ för försökspersoner över 5 år) och medelvärdet av HIV-1-RNA i plasma var 5,0 log10 kopior per ml. Mediandurationen vid prövningen var 10,1 månader för försökspersonerna som fick EPIVIR 150 mg plus RETROVIR och 9,2 månader för försökspersoner som fick didanosin som monoterapi. Resultaten sammanfattas i tabell 11.
Dosering en gång dagligen
ARROW (COL105677) var en 5-årig randomiserad multicenterstudie som utvärderade flera aspekter av klinisk hantering av HIV-1-infektion hos pediatriska försökspersoner. HIV-1-infekterade, behandlingsnaiva försökspersoner i åldern 3 månader till 17 år inkluderades och behandlades med en förstahandsbehandling innehållande EPIVIR 150 mg och abakavir, doserat två gånger dagligen enligt Världshälsoorganisationens rekommendationer. Efter minst 36 veckors behandling gavs försökspersonerna möjlighet att delta i randomisering 3 av ARROW-studien, där säkerheten och effekten av dosering en gång dagligen jämfördes med dosering två gånger dagligen av EPIVIR och abakavir, i kombination med ett tredje antiretroviralt medel. läkemedel i ytterligare 96 veckor. Av de 1 206 ursprungliga ARROW-personerna deltog 669 i randomisering 3. Virologisk suppression var inte ett krav för deltagande: vid baslinjen för randomisering 3 (efter minst 36 veckors behandling två gånger dagligen), 75 % av försökspersonerna två gånger dagligen kohorten var virologiskt undertryckta, jämfört med 71 % av försökspersonerna i kohorten en gång dagligen.
Andelen försökspersoner med HIV-1 RNA på mindre än 80 kopior per ml under 96 veckor visas i tabell 12. Skillnaderna mellan virologiska svar i de två behandlingsarmarna var jämförbara över baslinjeegenskaper för kön och ålder.
Analyser efter formulering visade att andelen försökspersoner med HIV-1 RNA på mindre än 80 kopior per ml vid randomisering och vecka 96 var högre hos försökspersoner som hade fått tablettformuleringar av EPIVIR 150 mg och abakavir (75 % [458/610] och 72 % [434/601]) än hos dem som hade fått lösningsformulering(er) (med EPIVIR 150 mg lösning givet i viktbandsbaserade doser på ungefär 8 mg per kg per dag) någon gång (52 % [29/56] och 54 % [30/56]), respektive [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. Dessa skillnader observerades i varje utvärderad åldersgrupp.
PATIENTINFORMATION
EPIVIR (EP-i-veer) (lamivudin) tabletter
EPIVIR (EP-i-veer) (lamivudin) oral lösning
Vilken är den viktigaste informationen jag borde veta om EPIVIR 150mg?
EPIVIR kan orsaka allvarliga biverkningar, inklusive:
- Försämring av hepatit B-virus hos personer som har HIV-1-infektion. Om du har HIV-1 (humant immunbristvirus typ 1) och hepatit B-virus (HBV)-infektion, kan ditt HBV förvärras (uppblåsning) om du slutar ta EPIVIR. En "flare-up" är när din HBV-infektion plötsligt kommer tillbaka på ett värre sätt än tidigare. Förvärrade leversjukdomar kan vara allvarliga och kan leda till döden.
- Får inte slut på EPIVIR. Fyll på ditt recept eller prata med din vårdgivare innan din EPIVIR är borta.
- Sluta inte med EPIVIR utan att först prata med din läkare.
- Om du slutar ta EPIVIR kommer din läkare att behöva kontrollera din hälsa ofta och ta blodprover regelbundet i flera månader för att kontrollera din lever.
- Resistent hepatit B-virus (HBV). Om du har HIV-1 och hepatit B kan hepatit B-viruset förändras (mutera) under din behandling med EPIVIR 150 mg och bli svårare att behandla (resistent).
- Använd med interferon- och ribavirinbaserade regimer. Försämring av leversjukdom som har orsakat dödsfall har inträffat hos personer infekterade med både HIV-1 och hepatit C-virus som tar antiretrovirala läkemedel och som också behandlas för hepatit C med interferon med eller utan ribavirin. Om du tar EPIVIR 150 mg och interferon med eller utan ribavirin, berätta för din läkare om du har några nya symtom.
Vad är EPIVIR?
EPIVIR är ett receptbelagt läkemedel som används tillsammans med andra antiretrovirala läkemedel för att behandla infektion med humant immunbristvirus (HIV-1).
HIV-1 är viruset som orsakar förvärvat immunbristsyndrom (AIDS).
EPIVIR tabletter och oral lösning (används för att behandla HIV-1-infektion) innehåller en högre dos av samma aktiva ingrediens (lamivudin) än vad som finns i läkemedlet EPIVIR-HBV tabletter och oral lösning (används för att behandla HBV). Om du har både HIV-1 och HBV ska du inte använda EPIVIR-HBV för att behandla dina infektioner.
Säkerheten och effekten av EPIVIR har inte fastställts hos barn under 3 månaders ålder.
Vem bör inte ta EPIVIR 150mg?
Ta inte EPIVIR om du är allergisk mot lamivudin eller något av innehållsämnena i EPIVIR. Se slutet av denna patientinformationsbroschyr för en komplett lista över ingredienser i EPIVIR.
Vad ska jag berätta för min vårdgivare innan jag tar EPIVIR?
Innan du tar EPIVIR 150 mg, berätta för din läkare om du:
- har eller har haft leverproblem, inklusive hepatit B- eller C-virusinfektion.
- har njurproblem.
- har diabetes. Varje 15 ml dos (150 mg) av EPIVIR oral lösning innehåller 3 gram sackaros.
- är gravid eller planerar att bli gravid. Att ta EPIVIR under graviditet har inte associerats med en ökad risk för fosterskador. Prata med din vårdgivare om du är gravid eller planerar att bli gravid. Graviditetsregistret. Det finns ett graviditetsregister för kvinnor som tar antiretrovirala läkemedel under graviditeten. Syftet med detta register är att samla in information om hälsan hos dig och ditt barn. Prata med din vårdgivare om hur du kan ta del av detta register.
- ammar eller planerar att amma. Amma inte om du tar EPIVIR.
- Du bör inte amma om du har HIV-1 på grund av risken att överföra HIV-1 till ditt barn.
Berätta för din vårdgivare om alla mediciner du tar, inklusive receptbelagda och receptfria läkemedel, vitaminer och växtbaserade kosttillskott.
Vissa läkemedel interagerar med EPIVIR. Håll en lista över dina läkemedel och visa den för din vårdgivare och apotekspersonal när du får ett nytt läkemedel. Du kan be din vårdgivare eller apotekspersonal om en lista över läkemedel som interagerar med EPIVIR.
Börja inte ta ett nytt läkemedel utan att berätta för din vårdgivare. Din vårdgivare kan berätta för dig om det är säkert att ta EPIVIR tillsammans med andra läkemedel.
Hur ska jag ta EPIVIR?
- Ta EPIVIR exakt som din läkare säger åt dig att ta det.
- Om du missar en dos av EPIVIR 150 mg, ta den så snart du kommer ihåg det. Ta inte 2 doser samtidigt eller ta mer än vad din läkare säger att du ska ta.
- Stanna under vård av en vårdgivare under behandling med EPIVIR.
- EPIVIR kan tas med eller utan mat.
- För barn 3 månader och äldre kommer din vårdgivare att ordinera en dos av EPIVIR 150 mg baserat på ditt barns kroppsvikt.
- Berätta för din vårdgivare om du eller ditt barn har problem med att svälja tabletter. EPIVIR 150mg kommer också som en vätska (oral lösning).
- Får inte slut på EPIVIR. Viruset i ditt blod kan öka och viruset kan bli svårare att behandla. När ditt utbud börjar ta slut, få mer från din vårdgivare eller apotek.
- Om du tar för mycket EPIVIR 150 mg, ring din vårdgivare eller gå till närmaste akutmottagning direkt.
Vilka är de möjliga biverkningarna av EPIVIR 150mg?
- EPIVIR 150 mg kan orsaka allvarliga biverkningar inklusive:
- Se "Vad är den viktigaste informationen jag borde veta om EPIVIR 150mg?"
- Ansamling av en syra i ditt blod (laktacidos). Laktacidos kan inträffa hos vissa personer som tar EPIVIR. Laktacidos är en allvarlig medicinsk nödsituation som kan orsaka dödsfall. Ring din vårdgivare omedelbart om du får något av följande symtom som kan vara tecken på laktacidos:
- känner mig väldigt svag eller trött
- känna sig kall, särskilt i armar och ben
- ovanlig (inte normal) muskelsmärta
- känna sig yr eller yr i huvudet
- problem att andas
- har snabba eller oregelbundna hjärtslag
- ont i magen med illamående och kräkningar
- Allvarliga leverproblem kan hända hos personer som tar EPIVIR. I vissa fall kan dessa allvarliga leverproblem leda till döden. Din lever kan bli stor (hepatomegali) och du kan utveckla fett i levern (steatos). Ring din vårdgivare omedelbart om du får något av följande tecken eller symtom på leverproblem:
- din hud eller den vita delen av dina ögon blir gul (gulsot)
- aptitlöshet i flera dagar eller längre
- illamående
- mörk eller "tefärgad" urin
- smärta, värk eller ömhet på höger sida av magområdet
- ljusfärgad avföring (tarmrörelser)
Du kan vara mer benägen att få laktacidos eller allvarliga leverproblem om du är kvinna eller mycket överviktig (fetma).
- Risk för inflammation i bukspottkörteln (pankreatit). Barn kan löpa risk att utveckla pankreatit under behandling med EPIVIR om de:
- har tagit nukleosidanalogläkemedel
- har en historia av pankreatit i det förflutna
- har andra riskfaktorer för pankreatit
Ring din vårdgivare omedelbart om ditt barn utvecklar tecken och symtom på pankreatit inklusive svår smärta i övre magområdet, med eller utan illamående och kräkningar. Din vårdgivare kan säga till dig att sluta ge EPIVIR 150 mg till ditt barn om deras symtom och blodprovsresultat visar att ditt barn kan ha pankreatit.
- Förändringar i ditt immunförsvar (immunrekonstitutionssyndrom) kan hända när du börjar ta HIV-1-läkemedel. Ditt immunförsvar kan bli starkare och börja bekämpa infektioner som har varit gömda i din kropp under lång tid. Berätta genast för din vårdgivare om du börjar få nya symtom efter att du har börjat ta EPIVIR.
De vanligaste biverkningarna av EPIVIR 150 mg hos vuxna inkluderar:
- huvudvärk
- nasala tecken och symtom
- illamående
- diarre
- mår generellt inte bra
- trötthet
- hosta
De vanligaste biverkningarna av EPIVIR 150 mg hos barn är feber och hosta.
Berätta för din vårdgivare om du har någon biverkning som stör dig eller som inte försvinner.
Dessa är inte alla möjliga biverkningar av EPIVIR. Ring din läkare för medicinsk rådgivning om biverkningar. Du kan rapportera biverkningar till FDA på 1-800-FDA-1088.
Hur ska jag förvara EPIVIR?
- Förvara EPIVIR 150 mg tabletter och oral lösning i rumstemperatur mellan 68°F till 77°F (20°C till 25°C).
- Förvara flaskor med EPIVIR 150 mg oral lösning väl tillslutna.
Förvara EPIVIR och alla läkemedel utom räckhåll för barn.
Allmän information om säker och effektiv användning av EPIVIR.
Läkemedel skrivs ibland ut för andra ändamål än de som anges i en patientinformationsbroschyr. Använd inte EPIVIR för ett tillstånd som det inte ordinerats för. Ge inte EPIVIR till andra personer, även om de har samma symtom som du har. Det kan skada dem.
Du kan fråga din vårdgivare eller apotekspersonal om information om EPIVIR 150mg som är skriven för vårdpersonal.
För mer information, gå till www.viivhealthcare.com eller ring 1-877-844-8872.
Vilka är ingredienserna i EPIVIR 150mg?
Aktiv ingrediens: lamivudin
Inaktiva Ingredienser:
EPIVIR 150 mg skårade 150 mg filmdragerade tabletter: hypromellos, magnesiumstearat, mikrokristallin cellulosa, polyetylenglykol, polysorbat 80, natriumstärkelseglykolat och titandioxid.
EPIVIR 300 mg filmdragerade tabletter: svart järnoxid, hypromellos, magnesiumstearat, mikrokristallin cellulosa, polyetylenglykol, polysorbat 80, natriumstärkelseglykolat och titandioxid.
EPIVIR 150 mg oral lösning: konstgjorda jordgubbs- och bananaromer, citronsyra (vattenfri), metylparaben, propylenglykol, propylparaben, natriumcitrat (dihydrat) och sackaros (200 mg per ml).
Denna patientinformation har godkänts av US Food and Drug Administration.