Xeloda 500mg Capecitabine Användning, biverkningar och dosering. Pris i onlineapotek. Generiska läkemedel utan recept.
Vad är Xeloda 500mg och hur används det?
Xeloda är ett receptbelagt läkemedel som används för att behandla symtom på cancer som tjocktarmscancer, kolorektal cancer och bröstcancer. Xeloda kan användas ensamt eller tillsammans med andra läkemedel.
Xeloda tillhör en klass av läkemedel som kallas Antineoplastics, Antimetabolite.
Det är inte känt om Xeloda 500 mg är säkert och effektivt för barn.
Vilka är de möjliga biverkningarna av Xeloda 500mg?
Xeloda kan orsaka allvarliga biverkningar inklusive:
- feber över 100,5 grader,
- illamående,
- aptitlöshet,
- äter mycket mindre än vanligt,
- kräkningar (mer än en gång per 24 timmar),
- svår diarré (mer än 4 gånger per dag eller under natten),
- blåsor eller sår i munnen,
- rött eller svullet tandkött,
- problem med att svälja,
- smärta, ömhet, rodnad, svullnad, blåsor eller fjällande hud på händer eller fötter,
- känner mig väldigt törstig eller varm,
- att inte kunna kissa,
- kraftig svettning,
- varm och torr hud,
- bröstsmärtor eller tryck,
- ojämna hjärtslag,
- andnöd,
- svullnad eller snabb viktökning,
- smärtsam eller svår urinering,
- svullnad i dina fötter eller vrister,
- känner mig trött,
- andnöd,
- mörk urin,
- lerfärgad avföring,
- gulfärgning av huden eller ögonen (gulsot),
- feber eller andra influensasymptom,
- hosta,
- hudsår,
- blek hud,
- lätt att få blåmärken,
- ovanlig blödning,
- känner sig yr,
- snabb hjärtfrekvens,
- öm hals,
- svullnad i ansiktet eller tungan,
- brännande i ögonen, och
- hudsmärta som följs av röda eller lila utslag (särskilt ansiktet eller överkroppen) och orsakar blåsor och fjällning
Få medicinsk hjälp omedelbart om du har något av symtomen som anges ovan.
De vanligaste biverkningarna av Xeloda 500 mg inkluderar:
- magont,
- förstoppning,
- magbesvär,
- trött känsla,
- milda hudutslag och
- domningar eller stickningar i händer eller fötter
Tala om för läkaren om du har någon biverkning som stör dig eller som inte försvinner.
Dessa är inte alla möjliga biverkningar av Xeloda. För mer information, fråga din läkare eller apotekspersonal.
Ring din läkare för medicinsk rådgivning om biverkningar. Du kan rapportera biverkningar till FDA på 1-800-FDA-1088.
VARNING
XELODA-WARFARIN INTERAKTION
XELODA 500mg Warfarin Interaktion: Patienter som samtidigt får capecitabin och oralt kumarinderivat av antikoagulantia bör få sitt antikoagulantiasvar (INR eller protrombintid) övervakat ofta för att justera antikoagulantdosen därefter. En kliniskt viktig XELODA-Warfarin läkemedelsinteraktion demonstrerades i en klinisk farmakologisk prövning [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER och DRUG INTERAKTIONER]. Förändrade koagulationsparametrar och/eller blödning, inklusive dödsfall, har rapporterats hos patienter som tar XELODA 500 mg samtidigt med kumarinderivativa antikoagulantia såsom warfarin och fenprokumon. Rapporter efter marknadsföring har visat kliniskt signifikanta ökningar av protrombintid (PT) och INR hos patienter som stabiliserades på antikoagulantia vid den tidpunkt då XELODA 500 mg introducerades. Dessa händelser inträffade inom flera dagar och upp till flera månader efter påbörjad behandling med XELODA 500 mg och, i några få fall, inom 1 månad efter avslutad XELODA. Dessa händelser inträffade hos patienter med och utan levermetastaser. Ålder över 60 och en cancerdiagnos disponerar oberoende patienter för en ökad risk för koagulopati.
BESKRIVNING
XELODA (capecitabin) är ett fluoropyrimidinkarbamat med antineoplastisk aktivitet. Det är en oralt administrerad systemisk prodrug av 5'-deoxi-5-fluorouridin (5'-DFUR) som omvandlas till 5-fluorouracil.
Det kemiska namnet för capecitabin är 5'-deoxi-5-fluoro-N-[(pentyloxi)karbonyl]-cytidin och har en molekylvikt på 359,35. Capecitabin har följande strukturformel:
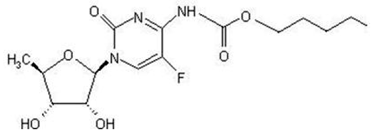
Capecitabin är ett vitt till benvitt kristallint pulver med en vattenlöslighet på 26 mg/ml vid 20°C.
XELODA tillhandahålls som bikonvexa, avlånga filmdragerade tabletter för oral administrering. Varje ljus persikofärgad tablett innehåller 150 mg capecitabin och varje persikofärgad tablett innehåller 500 mg capecitabin. De inaktiva ingredienserna i XELODA inkluderar: vattenfri laktos, kroskarmellosnatrium, hydroxipropylmetylcellulosa, mikrokristallin cellulosa, magnesiumstearat och renat vatten. Beläggningen av persika eller ljus persika innehåller hydroxipropylmetylcellulosa, talk, titandioxid och syntetiska gula och röda järnoxider.
INDIKATIONER
Kolorektal cancer
- XELODA är indicerat som ett enda läkemedel för adjuvant behandling hos patienter med Dukes C koloncancer som har genomgått fullständig resektion av den primära tumören när behandling med enbart fluoropyrimidinbehandling är att föredra. XELODA 500 mg var icke-sämre än 5fluorouracil och leukovorin (5-FU/LV) för sjukdomsfri överlevnad (DFS). Läkare bör överväga resultaten av kombinationskemoterapistudier, som har visat förbättringar i DFS och OS, när de förskriver XELODA 500 mg som enstaka medel vid adjuvansbehandling av Dukes C koloncancer.
- XELODA 500 mg är indicerat som förstahandsbehandling av patienter med metastaserande kolorektalt karcinom när behandling med enbart fluoropyrimidin är att föredra. Kombinationskemoterapi har visat en överlevnadsfördel jämfört med enbart 5-FU/LV. En överlevnadsfördel över 5-FU/LV har inte visats med XELODA 500 mg monoterapi. Användning av XELODA istället för 5FU/LV i kombinationer har inte studerats tillräckligt för att säkerställa säkerhet eller bevarande av överlevnadsfördelen.
Bröstcancer
- XELODA i kombination med docetaxel är indicerat för behandling av patienter med metastaserad bröstcancer efter misslyckande med tidigare antracyklininnehållande kemoterapi.
- XELODA 500 mg monoterapi är också indicerat för behandling av patienter med metastaserad bröstcancer som är resistenta mot både paklitaxel och en antracyklininnehållande kemoterapiregim eller resistenta mot paklitaxel och för vilka ytterligare antracyklinbehandling inte är indicerad (t.ex. patienter som har fått kumulativa doser på 400 mg/m2 doxorubicin eller doxorubicinekvivalenter). Resistens definieras som progressiv sjukdom under behandling, med eller utan ett initialt svar, eller återfall inom 6 månader efter avslutad behandling med en antracyklininnehållande adjuvant regim.
DOSERING OCH ADMINISTRERING
Viktiga administrationsinstruktioner
XELODA tabletter ska sväljas hela med vatten inom 30 minuter efter en måltid. XELODA 500mg är ett cellgifter. Följ tillämpliga särskilda hanterings- och kasseringsprocedurer.1 Om XELODA 500 mg tabletter måste skäras eller krossas, bör detta göras av en fackman som är utbildad i säker hantering av cellgifter med hjälp av lämplig utrustning och säkerhetsprocedurer. XELODA-dosen beräknas efter kroppsyta.
Standard startdos
Monoterapi (metastaserande kolorektal cancer, adjuvant kolorektal cancer, metastaserad bröstcancer)
Den rekommenderade dosen av XELODA är 1250 mg/m2 administrerat oralt två gånger dagligen (morgon och kväll; motsvarande 2500 mg/m2 total daglig dos) under 2 veckor följt av en 1-veckors viloperiod som ges som 3-veckorscykler (se ).
Adjuvansbehandling till patienter med Dukes C koloncancer rekommenderas i totalt 6 månader [dvs. XELODA 1250 mg/m2 oralt två gånger dagligen under 2 veckor följt av en 1-veckors viloperiod, givet som 3-veckorscykler för totalt på 8 cykler (24 veckor)].
I kombination med Docetaxel (metastaserande bröstcancer)
I kombination med docetaxel är den rekommenderade dosen av XELODA 1250 mg/m2 två gånger dagligen i 2 veckor följt av en veckas viloperiod, kombinerat med docetaxel vid 75 mg/m2 som en 1-timmes intravenös infusion var tredje vecka. Premedicinering, enligt docetaxelmärkningen, bör påbörjas före administrering av docetaxel för patienter som får kombinationen XELODA plus docetaxel. visar den totala dagliga dosen av XELODA efter kroppsyta och antalet tabletter som ska tas vid varje dos.
Riktlinjer för doshantering
Allmän
XELODA-dosering kan behöva individualiseras för att optimera patienthanteringen. Patienter bör övervakas noggrant med avseende på toxicitet och doser av XELODA 500 mg bör modifieras vid behov för att tillgodose individuell patienttolerans mot behandling [se Kliniska studier ]. Toxicitet på grund av administrering av XELODA 500 mg kan hanteras genom symptomatisk behandling, dosavbrott och justering av XELODA-dosen. När dosen väl har sänkts ska den inte ökas vid ett senare tillfälle. Doser av XELODA 500 mg utelämnade på grund av toxicitet ersätts eller återställs inte; istället bör patienten återuppta de planerade behandlingscyklerna.
Dosen av fenytoin och dosen av kumarinderivat antikoagulantia kan behöva minskas när något av läkemedlen ges samtidigt med XELODA [se LÄKEMEDELSINTERAKTIONER ].
Monoterapi (metastaserande kolorektal cancer, adjuvant kolorektal cancer, metastaserad bröstcancer)
XELODA dosändringsschema som beskrivs nedan (se ) rekommenderas för hantering av biverkningar.
I kombination med Docetaxel (metastaserande bröstcancer)
Dosändringar av XELODA 500 mg för toxicitet bör göras enligt ovan för XELODA. I början av en behandlingscykel, om en behandlingsfördröjning är indicerad för antingen XELODA 500 mg eller docetaxel, ska administreringen av båda medlen skjutas upp tills kraven för att återstarta båda läkemedlen är uppfyllda.
Dosreduktionsschemat för docetaxel när det används i kombination med XELODA för behandling av metastaserad bröstcancer visas i tabell 3.
Justering av startdos i speciella populationer
Nedsatt njurfunktion
Ingen justering av startdosen av XELODA 500 mg rekommenderas till patienter med lätt nedsatt njurfunktion (kreatininclearance = 51 till 80 ml/min [Cockroft och Gault, som visas nedan]). Hos patienter med måttligt nedsatt njurfunktion (baslinjekreatininclearance = 30 till 50 ml/min), en dosreduktion till 75 % av XELODA-startdosen när den används som monoterapi eller i kombination med docetaxel (från 1250 mg/m2 till 950 mg/m2) två gånger dagligen) rekommenderas [se Användning i specifika populationer och KLINISK FARMAKOLOGI ]. Efterföljande dosjustering rekommenderas enligt beskrivningen i och (beroende på regimen) om en patient utvecklar en biverkning av grad 2 till 4 [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]. Rekommendationerna för startdosjustering för patienter med måttligt nedsatt njurfunktion gäller både XELODA 500 mg monoterapi och XELODA 500 mg i kombination med docetaxel.
Cockroft och Gault Ekvation:
Geriatri
Läkare bör vara försiktiga när de övervakar effekterna av XELODA 500 mg hos äldre. Det finns inte tillräckligt med data för att ge en dosrekommendation.
HUR LEVERERAS
Doseringsformer Styrkor
XELODA tillhandahålls som bikonvexa, avlånga filmdragerade tabletter för oral administrering. Varje ljus persikofärgad tablett innehåller 150 mg capecitabin och varje persikofärgad tablett innehåller 500 mg capecitabin.
150 mg
Färg: Ljus persika Gravyr: XELODA på ena sidan och 150 på den andra 150 mg tabletter är förpackade i flaskor med 60 ( NDC 0004-1100-20), individuellt förpackade i en kartong.
500 mg
Färg: Persika Gravyr: XELODA 500 mg på ena sidan och 500 på den andra 500 mg tabletter är förpackade i flaskor med 120 ( NDC 0004-1101-50), individuellt förpackade i en kartong.
Förvaring Och Hantering
Förvara vid 25°C (77°F); utflykter tillåtna till 15° till 30°C (59° till 86°F). [Se USP-kontrollerad rumstemperatur]. HÅLL TÄT STÄNGD.
XELODA är ett cellgifter. Följ tillämpliga speciella hanterings- och kasseringsprocedurer.1 Alla oanvända produkter ska kasseras i enlighet med lokala krav eller program för återtagande av läkemedel.
REFERENSER
1. "OSHA farliga droger." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Distribueras av: Genentech USA, Inc. Medlem av Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Reviderad: maj 2021
BIEFFEKTER
Eftersom kliniska prövningar genomförs under vitt skilda förhållanden, kan biverkningsfrekvenser som observerats i de kliniska prövningarna av ett läkemedel inte direkt jämföras med frekvenser i kliniska prövningar av ett annat läkemedel och återspeglar kanske inte de frekvenser som observerats i praktiken.
Adjuvant tjocktarmscancer
visar de biverkningar som uppträdde hos ≥5 % av patienterna från en fas 3-studie hos patienter med Dukes C koloncancer som fick minst en dos studiemedicin och hade minst en säkerhetsbedömning. Totalt 995 patienter behandlades med 1250 mg/m2 två gånger om dagen av XELODA 500 mg administrerat i 2 veckor följt av en 1 veckas viloperiod, och 974 patienter fick 5-FU och leukovorin (20 mg/m2 leukovorin IV följt av 425 mg/m2 IV bolus 5FU dag 1-5 var 28:e dag). Mediandurationen av behandlingen var 164 dagar för capecitabinetbehandlade patienter och 145 dagar för 5-FU/LV-behandlade patienter. Totalt 112 (11 %) och 73 (7 %) capecitabin- respektive 5-FU/LV-behandlade patienter avbröt behandlingen på grund av biverkningar. Totalt 18 dödsfall på grund av alla orsaker inträffade antingen i studien eller inom 28 dagar efter att de fått studieläkemedlet: 8 (0,8 %) patienter randomiserades till XELODA 500 mg och 10 (1,0 %) randomiserades till 5-FU/LV.
visar laboratorieavvikelser av grad 3/4 som förekommer hos ≥1 % av patienterna från en fas 3-studie hos patienter med Dukes C koloncancer som fick minst en dos studiemedicin och hade minst en säkerhetsbedömning.
Metastaserande kolorektal cancer
Monoterapi
visar de biverkningar som uppträder hos ≥5 % av patienterna från att slå samman de två fas 3-studierna i första linjens metastaserande kolorektal cancer. Totalt 596 patienter med metastaserad kolorektal cancer behandlades med 1250 mg/m2 två gånger om dagen av XELODA administrerat under 2 veckor följt av en 1 veckas viloperiod, och 593 patienter fick 5-FU och leukovorin i Mayo-regimen (20 mg/m2 leucovorin IV följt av 425 mg/m2 IV bolus 5-FU, dag 1-5, var 28:e dag). I den poolade kolorektala databasen var medianbehandlingstiden 139 dagar för capecitabinbehandlade patienter och 140 dagar för 5-FU/LV-behandlade patienter. Totalt 78 (13%) och 63 (11%) capecitabin- respektive 5-FU/LV-behandlade patienter avbröt behandlingen på grund av biverkningar/interkurrent sjukdom. Totalt 82 dödsfall på grund av alla orsaker inträffade antingen i studien eller inom 28 dagar efter att de fått studieläkemedlet: 50 (8,4 %) patienter randomiserades till XELODA 500 mg och 32 (5,4 %) randomiserades till 5-FU/LV.
Bröstcancer
I kombination med Docetaxel
Följande data visas för kombinationsstudien med XELODA 500 mg och docetaxel hos patienter med metastaserad bröstcancer i och . I kombinationsarmen XELODA och docetaxel var behandlingen XELODA administrerad oralt 1250 mg/m2 två gånger dagligen som intermittent behandling (2 veckors behandling följt av 1 vecka utan behandling) i minst 6 veckor och docetaxel administrerat som en 1-timmes intravenös infusion kl. en dos på 75 mg/m2 den första dagen i varje 3-veckorscykel i minst 6 veckor. I monoterapiarmen administrerades docetaxel som en 1-timmes intravenös infusion i en dos på 100 mg/m2 den första dagen av varje 3-veckorscykel under minst 6 veckor. Medeldurationen av behandlingen var 129 dagar i kombinationsarmen och 98 dagar i monoterapiarmen. Totalt 66 patienter (26 %) i kombinationsarmen och 49 (19 %) i monoterapiarmen drog sig ur studien på grund av biverkningar. Andelen patienter som krävde dosreduktioner på grund av biverkningar var 65 % i kombinationsarmen och 36 % i monoterapiarmen. Andelen patienter som behövde avbryta behandlingen på grund av biverkningar i kombinationsarmen var 79 %. Behandlingsavbrott var en del av dosjusteringsschemat för kombinationsterapiarmen men inte för de patienter som behandlades med docetaxel som monoterapi.
Monoterapi
Följande data visas för studien på stadium IV bröstcancerpatienter som fick en dos på 1250 mg/m2 administrerad två gånger dagligen under 2 veckor följt av en 1 veckas viloperiod. Den genomsnittliga behandlingstiden var 114 dagar. Totalt avbröt 13 av 162 patienter (8%) behandlingen på grund av biverkningar/interkurrent sjukdom.
Kliniskt relevanta biverkningar hos
Kliniskt relevanta biverkningar rapporterade hos
Monoterapi (metastaserande kolorektal cancer, adjuvant kolorektal cancer, metastaserad bröstcancer)
Gastrointestinala: utspänd buk, dysfagi, proctalgi, ascites (0,1 %), magsår (0,1 %), ileus (0,3 %), giftig utvidgning av tarmen, gastroenterit (0,1 %)
Hud och subkutan.: nagelstörning (0,1%), ökad svettning (0,1%), ljuskänslighetsreaktion (0,1%), hudsår, klåda, strålningsåterkallelsesyndrom (0,2%)
Allmän: bröstsmärtor (0,2%), influensaliknande sjukdom, värmevallningar, smärta (0,1%), heshet, irritabilitet, svårigheter att gå, törst, bröstmassa, kollaps, fibros (0,1%), blödning, ödem, sedering
Neurologiska: sömnlöshet, ataxi (0,5%), tremor, dysfasi, encefalopati (0,1%), onormal koordination, dysartri, medvetslöshet (0,2%), försämrad balans
Ämnesomsättning: ökad vikt, kakexi (0,4 %), hypertriglyceridemi (0,1 %), hypokalemi, hypomagnesemi
Öga: konjunktivit
Andningsvägar: hosta (0,1 %), näsblod (0,1 %), astma (0,2 %), hemoptys, andnöd (0,1 %), dyspné
Hjärt: takykardi (0,1%), bradykardi, förmaksflimmer, ventrikulära extrasystoler, extrasystoler, myokardit (0,1%), perikardutgjutning
Infektioner: laryngit (1,0%), bronkit (0,2%), lunginflammation (0,2%), bronkopneumoni (0,2%), keratokonjunktivit, sepsis (0,3%), svampinfektioner (inklusive candidiasis) (0,2%)
Muskuloskeletala: myalgi, skelettsmärta (0,1%), artrit (0,1%), muskelsvaghet
Blod och lymfatiska: leukopeni (0,2%), koagulationsstörning (0,1%), benmärgsdepression (0,1%), idiopatisk trombocytopeni purpura (1,0%), pancytopeni (0,1%)
Kärl: hypotoni (0,2%), hypertoni (0,1%), lymfödem (0,1%), lungemboli (0,2%), cerebrovaskulär olycka (0,1%)
Psykiatrisk: depression, förvirring (0,1 %)
Njur: nedsatt njurfunktion (0,6 %)
Öra: vertigo
Lever och gallvägar: leverfibros (0,1 %), hepatit (0,1 %), kolestatisk hepatit (0,1 %), onormala leverfunktionstester
Immunförsvar: läkemedelsöverkänslighet (0,1 %)
XELODA 500mg i kombination med docetaxel (metastaserande bröstcancer)
Gastrointestinala: ileus (0,4 %), nekrotiserande enterokolit (0,4 %), esofagusår (0,4 %), hemorragisk diarré (0,8 %)
Neurologiska: ataxi (0,4 %), synkope (1,2 %), smakförlust (0,8 %), polyneuropati (0,4 %), migrän (0,4 %).
Hjärt: supraventrikulär takykardi (0,4 %)
Infektion: neutropen sepsis (2,4%), sepsis (0,4%), bronkopneumoni (0,4%) Blod &
Lymfatisk: agranulocytos (0,4%), protrombin minskat (0,4%)
Kärl: hypotoni (1,2%), venös flebit och tromboflebit (0,4%), postural hypotoni (0,8%)
Njur: njursvikt (0,4 %)
Lever och gallvägar: gulsot (0,4%), onormala leverfunktionstester (0,4%), leversvikt (0,4%), leverkoma (0,4%), levertoxicitet (0,4%)
Immunförsvar: överkänslighet (1,2 %)
Erfarenhet efter marknadsföring
Följande biverkningar har observerats efter marknadsföring: angioödem, leversvikt, tårkanalstenos, akut njursvikt sekundärt till uttorkning inklusive dödlig utgång [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ], kutan lupus erythematosus, hornhinnestörningar inklusive keratit, toxisk leukoencefalopati, allvarliga hudreaktioner som Stevens-Johnsons syndrom och toxisk epidermal nekrolys (TEN) [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ], ihållande eller allvarligt hand- och fotsyndrom kan så småningom leda till förlust av fingeravtryck [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ]
I fall av exponering för krossade XELODA 500 mg tabletter har följande biverkningar rapporterats: ögonirritation och svullnad, hudutslag, diarré, parestesi, huvudvärk, magirritation, kräkningar och illamående.
LÄKEMEDELSINTERAKTIONER
Läkemedelsinteraktioner
Antikoagulantia
Förändrade koagulationsparametrar och/eller blödning har rapporterats hos patienter som tar XELODA 500 mg samtidigt med antikoagulantia från kumarinderivat såsom warfarin och fenprokumon [se LÅDA VARNING ]. Dessa händelser inträffade inom flera dagar och upp till flera månader efter påbörjad behandling med XELODA 500 mg och, i några få fall, inom 1 månad efter avslutad XELODA. Dessa händelser inträffade hos patienter med och utan levermetastaser. I en läkemedelsinteraktionsstudie med administrering av engångsdos warfarin, var det en signifikant ökning av medel-AUC för S-warfarin [se KLINISK FARMAKOLOGI ]. Det maximala observerade INR-värdet ökade med 91 %. Denna interaktion beror troligen på en hämning av cytokrom P450 2C9 av capecitabin och/eller dess metaboliter.
fenytoin
Nivån av fenytoin bör övervakas noggrant hos patienter som tar XELODA 500 mg och fenytoindosen kan behöva minskas [se DOSERING OCH ADMINISTRERING ]. Rapporter efter marknadsföring tyder på att vissa patienter som fick XELODA 500 mg och fenytoin hade toxicitet i samband med förhöjda fenytoinnivåer. Formella läkemedelsinteraktionsstudier med fenytoin har inte utförts, men interaktionsmekanismen antas vara hämning av isoenzymet CYP2C9 av capecitabin och/eller dess metaboliter.
Leucovorin
Koncentrationen av 5-fluorouracil ökar och dess toxicitet kan förstärkas av leukovorin. Dödsfall från svår enterokolit, diarré och uttorkning har rapporterats hos äldre patienter som fått leukovorin och fluorouracil varje vecka.
CYP2C9 substrat
Förutom warfarin har inga formella läkemedelsinteraktionsstudier mellan XELODA och andra CYP2C9-substrat utförts. Försiktighet bör iakttas när XELODA administreras samtidigt med CYP2C9-substrat.
Allopurinol
Samtidig användning med allopurinol kan minska koncentrationen av capecitabins aktiva metaboliter [se KLINISK FARMAKOLOGI ], vilket kan minska XELODA-effekten. Undvik användningen av allopurinol under behandling med XELODA.
Interaktion mellan droger och livsmedel
Mat visade sig minska både hastigheten och graden av absorption av capecitabin [se KLINISK FARMAKOLOGI ]. I alla kliniska prövningar instruerades patienterna att administrera XELODA 500 mg inom 30 minuter efter en måltid. Det rekommenderas att XELODA administreras tillsammans med mat [se DOSERING OCH ADMINISTRERING ].
VARNINGAR
Ingår som en del av "FÖRSIKTIGHETSÅTGÄRDER" Sektion
FÖRSIKTIGHETSÅTGÄRDER
Koagulopati
Patienter som samtidigt behandlas med capecitabin och oralt kumarinderivat av antikoagulantia bör få sitt antikoagulantiasvar (INR eller protrombintid) övervakat noggrant med stor frekvens och antikoagulantia bör justeras därefter [se LÅDA VARNING och LÄKEMEDELSINTERAKTIONER ].
Diarre
XELODA 500 mg kan framkalla diarré, ibland allvarlig. Patienter med svår diarré bör övervakas noggrant och ges vätske- och elektrolytersättning om de blir uttorkade. Hos 875 patienter med antingen metastaserad bröstcancer eller kolorektal cancer som fick XELODA monoterapi, var mediantiden till första förekomsten av grad 2 till 4 diarré 34 dagar (intervall från 1 till 369 dagar). Mediandurationen av diarré grad 3 till 4 var 5 dagar. National Cancer Institute of Canada (NCIC) grad 2 diarré definieras som en ökning med 4 till 6 avföring/dag eller nattlig avföring, grad 3 diarré som en ökning med 7 till 9 avföring/dag eller inkontinens och malabsorption, och grad 4 diarré som en ökning med ≥10 avföring/dag eller kraftigt blodig diarré eller behov av parenteralt stöd. Om grad 2, 3 eller 4 diarré uppstår, ska administreringen av XELODA omedelbart avbrytas tills diarrén försvinner eller minskar i intensitet till grad 1 [se DOSERING OCH ADMINISTRERING ]. Standardbehandlingar mot diarré (t.ex. loperamid) rekommenderas.
Nekrotiserande enterokolit (tyflit) har rapporterats.
Kardiotoxicitet
Kardiotoxiciteten som observerats med XELODA 500 mg inkluderar hjärtinfarkt/ischemi, angina, rytmrubbningar, hjärtstillestånd, hjärtsvikt, plötslig död, elektrokardiografiska förändringar och kardiomyopati. Dessa biverkningar kan vara vanligare hos patienter med kranskärlssjukdom i anamnesen.
Dihydropyrimidindehydrogenasbrist
Baserat på rapporter efter marknadsföring löper patienter med vissa homozygota eller vissa sammansatta heterozygota mutationer i DPD-genen som resulterar i fullständig eller nästan fullständig frånvaro av DPD-aktivitet en ökad risk för akut tidig debut av toxicitet och allvarliga, livshotande eller dödliga biverkningar. reaktioner orsakade av XELODA (t.ex. mukosit, diarré, neutropeni och neurotoxicitet). Patienter med partiell DPD-aktivitet kan också ha ökad risk för allvarliga, livshotande eller dödliga biverkningar orsakade av XELODA.
Håll ut eller avbryt XELODA 500 mg permanent baserat på klinisk bedömning av början, varaktigheten och svårighetsgraden av de observerade toxiciteterna hos patienter med tecken på akut tidig debut eller ovanligt allvarlig toxicitet, vilket kan indikera nästan fullständig eller total frånvaro av DPD-aktivitet. Ingen XELODA-dos har visat sig vara säker för patienter med fullständig frånvaro av DPD-aktivitet. Det finns otillräckliga data för att rekommendera en specifik dos till patienter med partiell DPD-aktivitet, mätt med något specifikt test.
Uttorkning och njursvikt
Uttorkning har observerats och kan orsaka akut njursvikt som kan vara dödlig. Patienter med redan existerande nedsatt njurfunktion eller som får XELODA samtidigt med kända nefrotoxiska medel löper högre risk. Patienter med anorexi, asteni, illamående, kräkningar eller diarré kan snabbt bli uttorkade. Övervaka patienter när XELODA 500 mg administreras för att förebygga och korrigera uttorkning i början. Om uttorkning av grad 2 (eller högre) inträffar ska XELODA-behandlingen omedelbart avbrytas och uttorkningen korrigeras. Behandlingen ska inte återupptas förrän patienten är rehydrerad och eventuella utlösande orsaker har korrigerats eller kontrollerats. Dosjusteringar bör göras för den utlösande biverkningen vid behov [se DOSERING OCH ADMINISTRERING ].
Patienter med måttligt nedsatt njurfunktion vid baseline kräver dosreduktion [se DOSERING OCH ADMINISTRERING ]. Patienter med lätt och måttligt nedsatt njurfunktion vid baseline bör övervakas noggrant med avseende på biverkningar. Omedelbart avbrytande av behandlingen med efterföljande dosjusteringar rekommenderas om en patient utvecklar en biverkning av grad 2 till 4 enligt beskrivningen i [se DOSERING OCH ADMINISTRERING , Användning i specifika populationer , och KLINISK FARMAKOLOGI ].
Embryo-fetal toxicitet
Baserat på resultat från reproduktionsstudier på djur och dess verkningsmekanism, kan XELODA orsaka fosterskada när det ges till en gravid kvinna [se KLINISK FARMAKOLOGI ]. Begränsade tillgängliga data är inte tillräckliga för att informera om användning av XELODA 500 mg hos gravida kvinnor. I reproduktionsstudier på djur orsakade administrering av capecitabin till dräktiga djur under organogenesperioden embryodödlighet och teratogenicitet hos möss och embryodödlighet hos apor vid 0,2 respektive 0,6 gånger exponeringen (AUC) hos patienter som fick den rekommenderade dosen [se Användning i specifika populationer ]. Informera gravida kvinnor om den potentiella risken för ett foster. Informera kvinnor med reproduktionspotential att använda effektiva preventivmedel under behandlingen och i 6 månader efter den sista dosen av XELODA [se Användning i specifika populationer ].
Mukokutan och dermatologisk toxicitet
Allvarliga mukokutana reaktioner, vissa med dödlig utgång, såsom Stevens-Johnsons syndrom och Toxic Epidermal Necrolysis (TEN) kan förekomma hos patienter som behandlas med XELODA [se NEGATIVA REAKTIONER ]. XELODA 500mg ska avbrytas permanent hos patienter som upplever en allvarlig mukokutan reaktion som möjligen kan tillskrivas XELODA-behandling.
Hand- och fotsyndrom (palmar-plantar erytrodysestesi eller kemoterapiinducerat akralt erytem) är en kutan toxicitet. Mediantiden till debut var 79 dagar (från 11 till 360 dagar) med ett allvarlighetsintervall av grad 1 till 3 för patienter som fick XELODA 500 mg monoterapi i metastaserande miljö. Grad 1 kännetecknas av något av följande: domningar, dysestesi/parestesi, stickningar, smärtfri svullnad eller erytem i händer och/eller fötter och/eller obehag som inte stör normala aktiviteter. Grad 2 hand- och fotsyndrom definieras som smärtsamt erytem och svullnad av händer och/eller fötter och/eller obehag som påverkar patientens dagliga aktiviteter. Grad 3 hand- och fotsyndrom definieras som fuktig avskalning, sårbildning, blåsor eller svår smärta i händer och/eller fötter och/eller svåra obehag som gör att patienten inte kan arbeta eller utföra dagliga aktiviteter. Ihållande eller allvarligt hand- och fotsyndrom (grad 2 och högre) kan så småningom leda till förlust av fingeravtryck vilket kan påverka patientens identifiering. Om hand- och fotsyndrom grad 2 eller 3 uppträder, bör administreringen av XELODA avbrytas tills händelsen försvinner eller minskar i intensitet till grad 1. Efter hand- och fotsyndrom grad 3 bör efterföljande doser av XELODA 500 mg minskas [ ser DOSERING OCH ADMINISTRERING ].
Hyperbilirubinemi
Hos 875 patienter med antingen metastaserad bröstcancer eller kolorektal cancer som fick minst en dos av XELODA 1250 mg/m2 två gånger dagligen som monoterapi under 2 veckor följt av en veckas viloperiod, inträffade grad 3 (1,5-3 x ULN) hyperbilirubinemi i 15,2 % (n=133) av patienterna och hyperbilirubinemi grad 4 (>3 x ULN) förekom hos 3,9 % (n=34) av patienterna. Av 566 patienter som hade levermetastaser vid baslinjen och 309 patienter utan levermetastaser vid baslinjen, inträffade grad 3 eller 4 hyperbilirubinemi hos 22,8 % respektive 12,3 %. Av de 167 patienterna med grad 3 eller 4 hyperbilirubinemi hade 18,6 % (n=31) även postbaseline-förhöjningar (grad 1 till 4, utan förhöjningar vid baseline) i alkaliskt fosfatas och 27,5% (n=46) hade postbaseline-förhöjningar av transaminaser kl. när som helst (inte nödvändigtvis samtidigt). Majoriteten av dessa patienter, 64,5 % (n=20) och 71,7 % (n=33), hade levermetastaser vid baslinjen. Dessutom hade 57,5 % (n=96) och 35,3 % (n=59) av de 167 patienterna förhöjningar (grad 1 till 4) vid både prebaseline och postbaseline i alkaliskt fosfatas respektive transaminaser. Endast 7,8 % (n=13) och 3,0 % (n=5) hade grad 3 eller 4 förhöjda alkaliska fosfataser eller transaminaser.
Hos de 596 patienter som behandlades med XELODA 500 mg som förstahandsbehandling för metastaserad kolorektal cancer, var incidensen av grad 3 eller 4 hyperbilirubinemi liknande den övergripande säkerhetsdatabasen för kliniska prövningar av XELODA 500 mg monoterapi. Mediantiden till debut för grad 3 eller 4 hyperbilirubinemi i populationen av kolorektal cancer var 64 dagar och medianvärdet för totalbilirubin ökade från 8 μm/L vid baslinjen till 13 μm/L under behandling med XELODA. Av de 136 kolorektalcancerpatienterna med grad 3 eller 4 hyperbilirubinemi, hade 49 patienter grad 3 eller 4 hyperbilirubinemi som senast uppmätta värde, varav 46 hade levermetastaser vid baslinjen.
Hos 251 patienter med metastaserad bröstcancer som fick en kombination av XELODA 500 mg och docetaxel, inträffade hyperbilirubinemi grad 3 (1,5 till 3 x ULN) hos 7 % (n=17) och grad 4 (>3 x ULN) hyperbilirubinemi hos 2 % (n=5).
Om läkemedelsrelaterade förhöjningar av bilirubin av grad 3 till 4 inträffar, bör administreringen av XELODA 500 mg omedelbart avbrytas tills hyperbilirubinemi minskar till ≤3,0 X ULN [se rekommenderade dosändringar under DOSERING OCH ADMINISTRERING ].
Hematologiska
Hos 875 patienter med antingen metastaserad bröstcancer eller kolorektal cancer som fick en dos på 1250 mg/m2 administrerad två gånger dagligen som monoterapi under 2 veckor följt av en veckas viloperiod, hade 3,2 %, 1,7 % och 2,4 % av patienterna grad 3 eller 4 neutropeni, trombocytopeni eller minskning av hemoglobin, respektive. Hos 251 patienter med metastaserad bröstcancer som fick en dos av XELODA i kombination med docetaxel hade 68 % neutropeni grad 3 eller 4, 2,8 % hade grad 3 eller 4 trombocytopeni och 9,6 % hade grad 3 eller 4 anemi.
Patienter med neutrofilantal på
Geriatriska patienter
Patienter ≥80 år kan uppleva en högre incidens av grad 3 eller 4 biverkningar. Hos 875 patienter med antingen metastaserad bröstcancer eller kolorektal cancer som fick XELODA 500 mg monoterapi, upplevde 62 % av de 21 patienterna ≥80 år som behandlades med XELODA 500 mg en behandlingsrelaterad grad 3 eller 4 biverkning: diarré hos 6 (28,6 %) , illamående hos 3 (14,3 %), hand- och fotsyndrom hos 3 (14,3 %) och kräkningar hos 2 (9,5 %) patienter. Bland de 10 patienterna 70 år och äldre (inga patienter var >80 år gamla) som behandlades med XELODA 500 mg i kombination med docetaxel upplevde 30 % (3 av 10) av patienterna grad 3 eller 4 diarré och stomatit, och 40 % (4 av 10) upplevde grad 3 hand- och fotsyndrom.
Bland de 67 patienter ≥60 år som får XELODA i kombination med docetaxel, förekomsten av behandlingsrelaterade biverkningar grad 3 eller 4, behandlingsrelaterade allvarliga biverkningar, utsättningar på grund av biverkningar, behandlingsavbrott på grund av biverkningar och behandling avbrott inom de två första behandlingscyklerna var högre än i patientgruppen
Hos 995 patienter som fick XELODA som adjuvant terapi för Dukes C tjocktarmscancer efter resektion av primärtumören upplevde 41 % av de 398 patienterna ≥65 år som behandlades med XELODA en behandlingsrelaterad grad 3 eller 4 biverkning: hand- och -fotssyndrom hos 75 (18,8 %), diarré hos 52 (13,1 %), stomatit hos 12 (3,0 %), neutropeni/granulocytopeni hos 11 (2,8 %), kräkningar hos 6 (1,5 %) och illamående hos 5 (1,3 %). %) patienter. Hos patienter ≥65 år (alla randomiserade populationer; capecitabin 188 patienter, 5-FU/LV 208 patienter) behandlade för Dukes C koloncancer efter resektion av primärtumören, riskkvoterna för sjukdomsfri överlevnad och total överlevnad för XELODA jämfört med 5-FU/LV var 1,01 (95 % KI 0,80 – 1,27) respektive 1,04 (95 % KI 0,79 – 1,37).
Leverinsufficiens
Patienter med mild till måttlig leverdysfunktion på grund av levermetastaser bör övervakas noggrant när XELODA administreras. Effekten av allvarlig leverdysfunktion på dispositionen av XELODA är inte känd [se Användning i specifika populationer och KLINISK FARMAKOLOGI ].
Kombination med andra droger
Användning av XELODA i kombination med irinotekan har inte studerats tillräckligt.
Information om patientrådgivning
Rekommendera patienten att läsa den FDA-godkända patientmärkningen ( PATIENTINFORMATION ).
Diarre
Informera patienter som upplever grad 2 diarré (en ökning med 4 till 6 avföring/dag eller nattlig avföring) eller mer eller som upplever svår blodig diarré med svår buksmärta och feber att sluta ta XELODA. Ge patienter råd om användning av antidiarrébehandlingar (t.ex. loperamid) för att hantera diarré [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Kardiotoxicitet
Informera patienter om risken för kardiotoxicitet och att omedelbart kontakta sin vårdgivare eller att gå till en akutmottagning för nystart av bröstsmärtor, andnöd, yrsel eller yrsel [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Dihydropyrimidindehydrogenasbrist
Rekommendera patienter att meddela sin vårdgivare om de har en känd DPD-brist. Informera patienter om de har fullständig eller nästan fullständig frånvaro av DPD-aktivitet, de löper en ökad risk för akut tidig debut av toxicitet och allvarliga, livshotande eller dödliga biverkningar orsakade av XELODA (t.ex. mukosit, diarré, neutropeni och neurotoxicitet) [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Uttorkning och njursvikt
Instruera patienter som upplever grad 2 eller högre uttorkning (IV-vätskor indikerade VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Viktiga administrationsinstruktioner
Rekommendera patienter att svälja XELODA 500 mg tabletter hela med vatten inom 30 minuter efter en måltid. Rekommendera patienter och vårdgivare att inte krossa eller skära XELODA-tabletter. Rådgör patienter om de inte kan svälja XELODA tabletter hela, att informera sin vårdgivare [se DOSERING OCH ADMINISTRERING ].
Överkänslighet och angioödem
Informera patienter om att XELODA kan orsaka allvarliga överkänslighetsreaktioner och angioödem. Rekommendera patienter som har känt överkänslighet mot capecitabin eller 5-fluorouracil att informera sin vårdgivare [se KONTRAINDIKATIONER ]. Instruera patienter som utvecklar överkänslighetsreaktioner eller mukokutana symtom (t.ex. urtikaria, hudutslag, erytem, klåda eller svullnad av ansikte, läppar, tunga eller svalg som gör det svårt att svälja eller andas) att sluta ta XELODA och omedelbart kontakta sin vårdgivare eller att gå till akuten. [ser NEGATIVA REAKTIONER ].
Illamående
Instruera patienter som upplever illamående grad 2 (matintaget minskat avsevärt men kan äta intermittent) eller högre att sluta ta XELODA 500 mg omedelbart och att kontakta sin vårdgivare för hantering av illamående [se NEGATIVA REAKTIONER ].

Kräkningar
Instruera patienter som upplever kräkningar av grad 2 (2 till 5 episoder under en 24-timmarsperiod) eller mer att sluta ta XELODA omedelbart och att kontakta sin vårdgivare för hantering av kräkningar [se NEGATIVA REAKTIONER ].
Hand-och-fots syndrom
Instruera patienter som upplever hand- och fotsyndrom grad 2 (smärtande erytem och svullnad av händer och/eller fötter och/eller obehag som påverkar patienternas dagliga aktiviteter) eller högre att sluta ta XELODA 500 mg omedelbart och kontakta sin vårdgivare . Informera patienterna om att initiering av symtomatisk behandling rekommenderas, och hand- och fotsyndrom kan leda till förlust av fingeravtryck vilket kan påverka personlig identifiering [se NEGATIVA REAKTIONER ].
Stomatit
Informera patienter som upplever grad 2 stomatit (smärtsamt erytem, ödem eller sår i munnen eller tungan, men kan äta) eller högre att sluta ta XELODA omedelbart och att kontakta sin vårdgivare [se NEGATIVA REAKTIONER ].
Feber Och Neutropeni
Informera patienter som utvecklar feber på 100,5°F eller högre eller andra tecken på potentiell infektion att kontakta sin vårdgivare [se NEGATIVA REAKTIONER ].
Embryo-fetal toxicitet
Informera kvinnor om reproduktionspotential om den potentiella risken för ett foster och att använda effektiva preventivmedel under behandling med XELODA 500 mg och i 6 månader efter den sista dosen. Rekommendera kvinnor att informera sin vårdgivare om en känd eller misstänkt graviditet [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , Användning i specifika populationer ].
Informera manliga patienter med kvinnliga partners med reproduktionspotential att använda effektiva preventivmedel under behandling med XELODA 500 mg och i 3 månader efter den sista dosen [se Användning i specifika populationer ].
Laktation
Avråda kvinnor att inte amma under behandling med XELODA 500 mg och i 2 veckor efter den sista dosen [se Användning i specifika populationer ].
Icke-klinisk toxikologi
Karcinogenes, Mutagenes, Nedsatt fertilitet
Adekvata studier som undersöker capecitabins karcinogena potential har inte utförts. Capecitabin var inte mutagent in vitro för bakterier (Ames-test) eller däggdjursceller (kinesisk hamster V79/HPRT-genmutationsanalys). Capecitabin var klastogent in vitro för humana perifera blodlymfocyter men inte klastogent in vivo för benmärg hos mus (mikronukleustest). Fluorouracil orsakar mutationer i bakterier och jäst. Fluorouracil orsakar också kromosomavvikelser i musmikronkärntestet in vivo.
studier av fertilitet och allmän reproduktion hos honmöss störde orala capecitabindoser på 760 mg/kg/dag (cirka 2300 mg/m2/dag) brunst och orsakade följaktligen en minskning av fertiliteten. Hos möss som blev gravida överlevde inga foster denna dos. Störningen i brunst var reversibel. Hos män orsakade denna dos degenerativa förändringar i testiklarna, inklusive minskningar av antalet spermatocyter och spermatider. I separata farmakokinetiska studier gav denna dos på möss 5'-DFUR AUC-värden omkring 0,7 gånger motsvarande värden hos patienter som fick den rekommenderade dagliga dosen.
Användning i specifika populationer
Graviditet
Risksammanfattning
Baserat på resultat i reproduktionsstudier på djur och dess verkningsmekanism, kan XELODA 500 mg orsaka fosterskada när det administreras till en gravid kvinna [se KLINISK FARMAKOLOGI ]. Begränsade tillgängliga humandata är inte tillräckliga för att informera om den läkemedelsrelaterade risken under graviditet. I reproduktionsstudier på djur orsakade administrering av capecitabin till dräktiga djur under organogenesperioden embryoletalitet och teratogenicitet hos möss och embryoletalitet hos apor vid 0,2 respektive 0,6 gånger exponeringen (AUC) hos patienter som fick den rekommenderade dosen [se]. Data ]. Informera gravida kvinnor om den potentiella risken för ett foster.
Den uppskattade bakgrundsrisken för allvarliga fosterskador och missfall för den indikerade populationen är okänd. I den allmänna befolkningen i USA är den uppskattade bakgrundsrisken för stora fosterskador och missfall i kliniskt erkända graviditeter 2-4% respektive 15-20%.
Data
Djurdata
Oral administrering av capecitabin till gravida möss under organogenesperioden vid en dos på 198 mg/kg/dag orsakade missbildningar och embryodödlighet. I separata farmakokinetiska studier gav denna dos på möss 5'-DFUR AUC-värden som var ungefär 0,2 gånger AUC-värdena hos patienter som fick den rekommenderade dagliga dosen. Missbildningar hos möss inkluderade gomspalt, anoftalmi, mikroftalmi, oligodaktyli, polydaktyli, syndaktyli, kinky svans och utvidgning av cerebrala ventriklar. Oral administrering av capecitabin till dräktiga apor under organogenesperioden i en dos på 90 mg/kg/dag orsakade fosterdödlighet. Denna dos gav 5'-DFUR AUC-värden som var ungefär 0,6 gånger AUC-värdena hos patienter som fick den rekommenderade dagliga dosen.
Laktation
Risksammanfattning
Det finns ingen information om förekomsten av capecitabin i modersmjölk, eller om dess effekter på mjölkproduktionen eller det ammade barnet. Capecitabin-metaboliter fanns i mjölken hos ammande möss [se Data ]. På grund av risken för allvarliga biverkningar från capecitabinexponering hos ammade spädbarn, råd kvinnor att inte amma under behandling med XELODA 500 mg och under 2 veckor efter den sista dosen.
Data
Ammande möss som fick en oral engångsdos capecitabin utsöndrade betydande mängder capecitabinmetaboliter i mjölken.
Kvinnor Och Hanar Av Reproduktionspotential
Graviditetstest
Graviditetstest rekommenderas för kvinnor med reproduktionspotential innan XELODA påbörjas.
Preventivmedel
Kvinnor
XELODA 500mg kan orsaka fosterskador när det administreras till en gravid kvinna [se Graviditet ]. Informera kvinnor med reproduktionspotential att använda effektiva preventivmedel under behandlingen och i 6 månader efter den sista dosen av XELODA.
Män
Baserat på upptäckter av genetisk toxicitet, råd manliga patienter med kvinnliga partner med reproduktionspotential att använda effektiva preventivmedel under behandlingen och i 3 månader efter den sista dosen av XELODA [se Icke-klinisk toxikologi ].
Infertilitet
Baserat på djurstudier kan XELODA försämra fertiliteten hos kvinnor och män med reproduktionspotential [se Icke-klinisk toxikologi ].
Pediatrisk användning
Säkerheten och effektiviteten av XELODA hos pediatriska patienter har inte fastställts. Ingen klinisk nytta visades i två enarmsstudier på pediatriska patienter med nydiagnostiserade hjärnstamgliom och höggradiga gliom. I båda studierna fick pediatriska patienter en pediatrisk prövningsformulering av capecitabin samtidigt med och efter avslutad strålbehandling (total dos på 5580 cGy i 180 cGy-fraktioner). Den relativa biotillgängligheten för undersökningsberedningen till XELODA 500 mg var liknande.
Den första studien genomfördes på 22 pediatriska patienter (medianålder 8 år, intervall 5-17 år) med nyligen diagnostiserade icke-spridade inre diffusa hjärnstamgliom och höggradiga gliom. I den dosfinnande delen av studien fick patienter capecitabin med samtidig strålbehandling i doser från 500 mg/m2 till 850 mg/m2 var 12:e timme i upp till 9 veckor. Efter en 2 veckors paus fick patienterna 1250 mg/m2 capecitabin var 12:e timme på dagarna 1-14 i en 21-dagars cykel i upp till 3 cykler. Den maximala tolererade dosen (MTD) av capecitabin administrerad samtidigt med strålbehandling var 650 mg/m2 var 12:e timme. De huvudsakliga dosbegränsande toxiciteterna var palmar-plantar erytrodysestesi och förhöjd alaninaminotransferas (ALAT).
Den andra studien genomfördes på 34 ytterligare pediatriska patienter med nyligen diagnostiserade icke-spridade inre diffusa hjärnstamgliom (medianålder 7 år, intervall 3-16 år) och 10 pediatriska patienter som fick MTD av capecitabin i den dosfinnande studien och uppfyllde behörighetskriterierna för denna prövning. Alla patienter fick 650 mg/m2 capecitabin var 12:e timme med samtidig strålbehandling i upp till 9 veckor. Efter en 2 veckors paus fick patienterna 1250 mg/m2 capecitabin var 12:e timme på dagarna 1-14 i en 21-dagarscykel i upp till 3 cykler.
Det fanns ingen förbättring i ett års progressionsfri överlevnadsfrekvens och ett års total överlevnad hos pediatriska patienter med nyligen diagnostiserade intrinsiska hjärnstamgliom som fick capecitabin i förhållande till en liknande population av pediatriska patienter som deltog i andra kliniska prövningar.
Biverkningsprofilen för capecitabin överensstämde med den kända biverkningsprofilen hos vuxna, med undantag för laboratorieavvikelser som förekom vanligare hos pediatriska patienter. De mest frekvent rapporterade laboratorieavvikelserna (incidens per patient ≥40%) var ökad ALAT (75%), lymfocytopeni (73%), leukopeni (73%), hypokalemi (68%), trombocytopeni (57%), hypoalbuminemi (55). %), neutropeni (50 %), låg hematokrit (50 %), hypokalcemi (48 %), hypofosfatemi (45 %) och hyponatremi (45 %).
Geriatrisk användning
Läkare bör vara särskilt uppmärksamma på att övervaka de skadliga effekterna av XELODA 500 mg hos äldre [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER ].
Leverinsufficiens
Iaktta försiktighet när patienter med mild till måttlig leverdysfunktion på grund av levermetastaser behandlas med XELODA. Effekten av allvarlig leverdysfunktion på XELODA är inte känd [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER och KLINISK FARMAKOLOGI ].
Njurinsufficiens
Patienter med måttligt (kreatininclearance = 30 till 50 ml/min) och allvarligt (kreatininclearance KONTRAINDIKATIONER , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , DOSERING OCH ADMINISTRERING , och KLINISK FARMAKOLOGI ].
ÖVERDOS
Symptomen på akut överdos skulle inkludera illamående, kräkningar, diarré, gastrointestinala irritationer och blödningar samt benmärgsdepression. Medicinsk hantering av överdosering bör inkludera sedvanliga stödjande medicinska insatser som syftar till att korrigera de kliniska manifestationerna. Även om ingen klinisk erfarenhet av att använda dialys som behandling för XELODA 500 mg överdosering har rapporterats, kan dialys vara till nytta för att minska cirkulerande koncentrationer av 5'-DFUR, en lågmolekylär metabolit av modersubstansen.
Engångsdoser av XELODA 500 mg var inte dödliga för möss, råttor och apor vid doser upp till 2000 mg/kg (2,4, 4,8 och 9,6 gånger den rekommenderade dagliga dosen för människor på mg/m2-basis).
KONTRAINDIKATIONER
Svårt nedsatt njurfunktion
XELODA är kontraindicerat hos patienter med gravt nedsatt njurfunktion (kreatininclearance under 30 ml/min [Cockroft och Gault]) [se Användning i specifika populationer och KLINISK FARMAKOLOGI ].
Överkänslighet
XELODA 500 mg är kontraindicerat till patienter med känd överkänslighet mot capecitabin eller mot någon av dess komponenter. XELODA är kontraindicerat till patienter som har en känd överkänslighet mot 5fluorouracil.
KLINISK FARMAKOLOGI
Handlingsmekanism
Enzymer omvandlar capecitabin till 5-fluorouracil (5-FU) in vivo. Både normala celler och tumörceller metaboliserar 5-FU till 5-fluor-2'-deoxiuridinmonofosfat (FdUMP) och 5-fluoruridintrifosfat (FUTP). Dessa metaboliter orsakar cellskador genom två olika mekanismer. Först binder FdUMP och folatkofaktorn, N5-10-metylentetrahydrofolat, till tymidylatsyntas (TS) för att bilda ett kovalent bundet ternärt komplex. Denna bindning hämmar bildningen av tymidylat från 2'-deoxiuridylat. Tymidylat är den nödvändiga prekursorn för tymidintrifosfat, som är väsentligt för syntesen av DNA, så att en brist på denna förening kan hämma celldelningen. För det andra kan nukleära transkriptionsenzymer felaktigt inkorporera FUTP i stället för uridintrifosfat (UTP) under syntesen av RNA. Detta metaboliska fel kan störa RNA-bearbetning och proteinsyntes.
Farmakokinetik
Absorption
Efter oral administrering av 1255 mg/m2 två gånger dagligen till cancerpatienter nådde capecitabin maximala blodnivåer inom cirka 1,5 timmar (Tmax) och toppnivåer av 5-FU inträffade något senare, efter 2 timmar. Mat minskade både absorptionshastigheten och omfattningen av capecitabin med genomsnittlig Cmax och AUC0-∞ minskade med 60 % respektive 35 %. Cmax och AUC0-∞ för 5-FU reducerades också med föda med 43 % respektive 21 %. Mat försenad Tmax för både förälder och 5-FU med 1,5 timmar [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , DOSERING OCH ADMINISTRERING , och Interaktion mellan droger och livsmedel ].
Farmakokinetiken för XELODA 500 mg och dess metaboliter har utvärderats hos cirka 200 cancerpatienter över ett dosintervall på 500 till 3500 mg/m2/dag. Över detta intervall var farmakokinetiken för XELODA och dess metabolit, 5'-DFCR, dosproportionella och förändrades inte över tiden. Ökningen av AUC för 5'-DFUR och 5-FU var dock större än proportionell mot dosökningen och AUC för 5-FU var 34 % högre på dag 14 än på dag 1. Variabiliteten mellan patienter i Cmax och AUC för 5-FU var större än 85 %.
Distribution
Plasmaproteinbindningen av capecitabin och dess metaboliter är mindre än 60 % och är inte koncentrationsberoende. Capecitabin var primärt bundet till humant albumin (ungefär 35%). XELODA 500 mg har en låg potential för farmakokinetiska interaktioner relaterade till plasmaproteinbindning.
Bioaktivering och metabolism
Capecitabin metaboliseras i stor utsträckning enzymatiskt till 5-FU. I levern hydrolyserar ett 60 kDa karboxyesteras mycket av föreningen till 5'-deoxi-5-fluorocytidin (5'-DFCR). Cytidindeaminas, ett enzym som finns i de flesta vävnader, inklusive tumörer, omvandlar därefter 5'-DFCR till 5'-DFUR. Enzymet, tymidinfosforylas (dThdPase), hydrolyserar sedan 5'DFUR till det aktiva läkemedlet 5-FU. Många vävnader i hela kroppen uttrycker tymidinfosforylas. Vissa humana karcinom uttrycker detta enzym i högre koncentrationer än omgivande normala vävnader. Efter oral administrering av XELODA 7 dagar före operation hos patienter med kolorektal cancer, var mediankvoten mellan 5-FU-koncentrationen i kolorektala tumörer och intilliggande vävnader 2,9 (intervall från 0,9 till 8,0). Dessa förhållanden har inte utvärderats hos bröstcancerpatienter eller jämfört med 5-FU-infusion.
Metabolisk väg för capecitabin till 5-FU
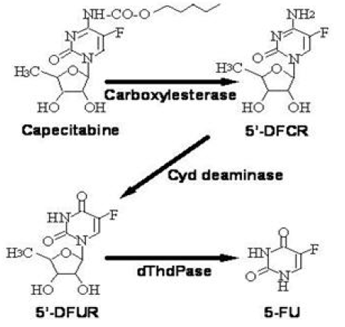
Enzymet dihydropyrimidindehydrogenas hydrerar 5-FU, produkten av capecitabinmetabolism, till det mycket mindre giftiga 5-fluoro-5, 6-dihydrofluorouracil (FUH2). Dihydropyrimidinas klyver pyrimidinringen för att ge 5-fluor-ureido-propionsyra (FUPA). Slutligen klyver β-ureido-propionas FUPA till α-fluoro-β-alanin (FBAL) som rensas ut i urinen.
Enzymatiska studier in vitro med humana levermikrosomer visade att capecitabin och dess metaboliter (5'-DFUR, 5'-DFCR, 5-FU och FBAL) inte hämmade metabolismen av testsubstrat av cytokrom P450 isoenzymer 1A2, 2A6, 3A4, 2C19, 2D6 och 2E1.
Exkretion
Capecitabin och dess metaboliter utsöndras huvudsakligen i urinen; 95,5 % av den administrerade capecitabindosen återfinns i urinen. Fekal utsöndring är minimal (2,6%). Den huvudsakliga metaboliten som utsöndras i urinen är FBAL som representerar 57 % av den administrerade dosen. Cirka 3 % av den administrerade dosen utsöndras i urinen som oförändrat läkemedel. Eliminationshalveringstiden för både moderkapecitabin och 5-FU var cirka 0,75 timmar.
Effekt av ålder, kön och ras på farmakokinetiken för capecitabin
En populationsanalys av sammanslagna data från de två stora kontrollerade studierna på patienter med metastaserad kolorektal cancer (n=505) som administrerades XELODA 500 mg vid 1250 mg/m2 två gånger om dagen visade att kön (202 kvinnor och 303 män) och ras (455 vita/kaukasiska patienter, 22 svarta patienter och 28 patienter av annan ras) har ingen inverkan på farmakokinetiken för 5'DFUR, 5-FU och FBAL. Ålder har ingen signifikant inverkan på farmakokinetiken för 5'-DFUR och 5-FU över intervallet 27 till 86 år. En 20% ökning i ålder resulterar i en 15% ökning av AUC för FBAL [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER och DOSERING OCH ADMINISTRERING ].
Efter oral administrering av 825 mg/m2 capecitabin två gånger dagligen under 14 dagar hade japanska patienter (n=18) cirka 36 % lägre Cmax och 24 % lägre AUC för capecitabin än de kaukasiska patienterna (n=22). Japanska patienter hade också cirka 25 % lägre Cmax och 34 % lägre AUC för FBAL än de kaukasiska patienterna. Den kliniska betydelsen av dessa skillnader är okänd. Inga signifikanta skillnader inträffade i exponeringen för andra metaboliter (5'-DFCR, 5'-DFUR och 5FU).
Effekt av leverinsufficiens
XELODA har utvärderats hos 13 patienter med mild till måttlig leverdysfunktion på grund av levermetastaser definierade av en sammansatt poäng inklusive bilirubin, ASAT/ALT och alkaliskt fosfatas efter en engångsdos på 1255 mg/m2 av XELODA. Både AUC0-∞ och Cmax för capecitabin ökade med 60 % hos patienter med leverdysfunktion jämfört med patienter med normal leverfunktion (n=14). AUC0-∞ och Cmax för 5-FU påverkades inte. Hos patienter med mild till måttlig leverdysfunktion på grund av levermetastaser, bör försiktighet iakttas när XELODA 500 mg administreras. Effekten av allvarlig leverdysfunktion på XELODA 500 mg är inte känd [se VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER och Användning i speciella populationer ].
Effekt av njurinsufficiens
Efter oral administrering av 1250 mg/m2 capecitabin två gånger dagligen till cancerpatienter med olika grader av nedsatt njurfunktion, visade patienter med måttligt (kreatininclearance = 30 till 50 ml/min) och allvarligt (kreatininclearance 80 ml/min). Systemisk exponering för 5'-DFUR var 42 % och 71 % högre hos patienter med måttligt respektive gravt nedsatt njurfunktion än hos normala patienter. Systemisk exponering för capecitabin var cirka 25 % högre hos både måttligt och gravt nedsatt njurfunktion [se DOSERING OCH ADMINISTRERING , KONTRAINDIKATIONER , VARNINGAR OCH FÖRSIKTIGHETSÅTGÄRDER , och Användning i speciella populationer ].
Effekt av capecitabin på farmakokinetiken för warfarin
Hos fyra patienter med cancer ökade kronisk administrering av capecitabin (1250 mg/m2 två gånger dagligen) med en enkeldos på 20 mg warfarin medel-AUC för S-warfarin med 57 % och minskade dess clearance med 37 %. Baslinjekorrigerad AUC för INR hos dessa 4 patienter ökade med 2,8 gånger och det maximala observerade medelvärdet för INR ökade med 91 % [se LÅDA VARNING och LÄKEMEDELSINTERAKTIONER ].
Effekt av antacida på farmakokinetiken för capecitabin
När Maalox® (20 mL), ett antacidum innehållande aluminiumhydroxid och magnesiumhydroxid, administrerades omedelbart efter XELODA (1250 mg/m2, n=12 cancerpatienter), ökade AUC och Cmax med 16 % respektive 35 %. för capecitabin och med 18 % respektive 22 % för 5'-DFCR. Ingen effekt observerades på de andra tre huvudmetaboliterna (5'-DFUR, 5-FU, FBAL) av XELODA.
Effekt av Allopurinol på Capecitabin
Publicerad litteratur rapporterade att samtidig användning med allopurinol kan minska omvandlingen av capecitabin till de aktiva metaboliterna, FdUMP och FUTP; den kliniska signifikansen karakteriserades dock inte helt.
Effekt av capecitabin på farmakokinetiken för docetaxel och vice versa
En fas 1-studie utvärderade effekten av XELODA på farmakokinetiken för docetaxel (Taxotere®) och effekten av docetaxel på farmakokinetiken för XELODA utfördes på 26 patienter med solida tumörer. XELODA visade sig inte ha någon effekt på farmakokinetiken för docetaxel (Cmax och AUC) och docetaxel har ingen effekt på farmakokinetiken för capecitabin och 5-FU-prekursorn 5'-DFUR.
Kliniska studier
Adjuvant tjocktarmscancer
En multicenter randomiserad, kontrollerad klinisk fas 3-studie på patienter med Dukes C koloncancer (X-ACT) gav data om användningen av XELODA för adjuvansbehandling av patienter med koloncancer. Det primära syftet med studien var att jämföra sjukdomsfri överlevnad (DFS) hos patienter som fick XELODA med de som enbart fick IV 5-FU/LV. I denna studie randomiserades 1987 patienter antingen till behandling med XELODA 1250 mg/m2 oralt två gånger dagligen under 2 veckor följt av en 1-veckors viloperiod, givet som 3-veckorscykler för totalt 8 cykler (24 veckor) eller IV. bolus 5-FU 425 mg/m2 och 20 mg/m2 IV leukovorin dag 1 till 5, givet som 4-veckorscykler under totalt 6 cykler (24 veckor). Patienterna i studien krävdes att vara mellan 18 och 75 år med histologiskt bekräftad Dukes tjocktarmscancer i stadium C med minst en positiv lymfkörtel och ha genomgått (inom 8 veckor före randomisering) fullständig resektion av primärtumören utan makroskopiska eller mikroskopiska bevis på kvarvarande tumör. Patienterna behövde inte heller ha någon tidigare cytotoxisk kemoterapi eller immunterapi (förutom steroider) och hade en ECOG-prestandastatus på 0 eller 1 (KPS ≥ 70 %), ANC ≥ 1,5x109/L, trombocyter ≥ 100x109/L, serumkreatinin ≤ 1,5 ULN, totalt bilirubin ≤ 1,5 ULN, AST/ALT ≤ 2,5 ULN och CEA inom normala gränser vid tidpunkten för randomisering.
Baslinjedemografin för XELODA- och 5-FU/LV-patienter visas i tabell 10. Baslinjeegenskaperna var välbalanserade mellan armarna.
Alla patienter med normal njurfunktion eller lätt nedsatt njurfunktion påbörjade behandlingen med full startdos på 1250 mg/m2 oralt två gånger dagligen. Startdosen reducerades hos patienter med måttligt nedsatt njurfunktion (beräknat kreatininclearance 30 till 50 ml/min) vid baslinjen [se DOSERING OCH ADMINISTRERING ]. Därefter justerades doserna för alla patienter vid behov enligt toxicitet. Doshantering för XELODA 500 mg inkluderade dosminskningar, cykelförseningar och behandlingsavbrott (se tabell 11).
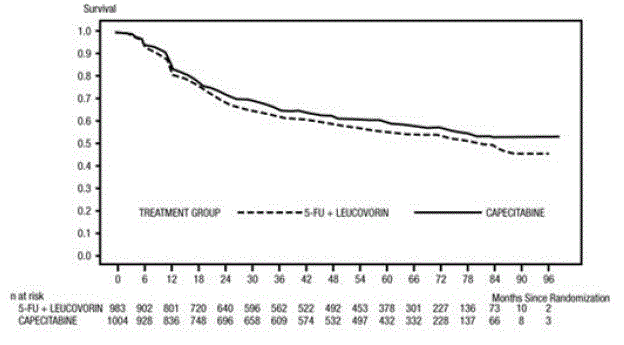
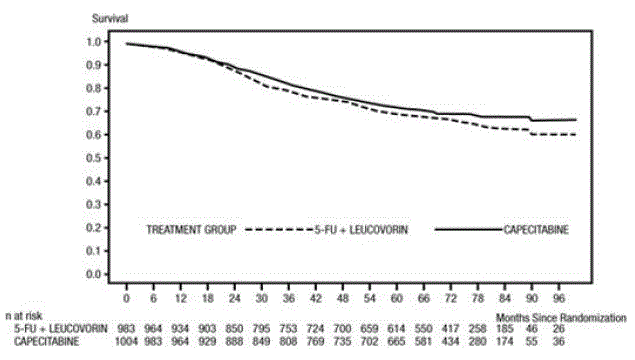
Medianuppföljningen vid analystillfället var 83 månader (6,9 år). Riskkvoten för DFS för XELODA 500 mg jämfört med 5-FU/LV var 0,88 (95 % KI 0,77 – 1,01) (se och Figur 1 ). Eftersom den övre 2-sidiga 95 % konfidensgränsen för hazard ratio var mindre än 1,20, var XELODA inte sämre än 5-FU/LV. Valet av non-inferiority-marginalen på 1,20 motsvarar bibehållandet av cirka 75 % av 5-FU/LV-effekten på DFS. Hazard ratio för XELODA jämfört med 5-FU/LV med avseende på total överlevnad var 0,86 (95 % KI 0,74 – 1,01). Den totala överlevnaden efter 5 år var 71,4 % för XELODA och 68,4 % för 5-FU/LV (se figur 2).
Figur 1 Kaplan-Meier uppskattningar av sjukdomsfri överlevnad (all randomiserad population)a
Figur 2 Kaplan-Meier uppskattningar av total överlevnad (all randomiserad population)
Metastaserande kolorektal cancer
Allmän
Den rekommenderade dosen av XELODA 500 mg bestämdes i en öppen, randomiserad klinisk studie, som undersökte effektiviteten och säkerheten av kontinuerlig behandling med capecitabin (1331 mg/m2/dag i två uppdelade doser, n=39), intermittent behandling med capecitabin ( 2510 mg/m2/dag i två uppdelade doser, n=34), och intermittent behandling med capecitabin i kombination med oralt leukovorin (LV) (capecitabin 1657 mg/m2/dag i två uppdelade doser, n=35; leukovorin 60 mg/dag dag) hos patienter med avancerat och/eller metastaserande kolorektalt karcinom i första linjens metastaserande miljö. Det fanns ingen uppenbar fördel i svarsfrekvensen för att lägga till leukovorin till XELODA; dock ökade toxiciteten. XELODA, 1250 mg/m2 två gånger dagligen under 14 dagar följt av en veckas vila, valdes ut för vidare klinisk utveckling baserat på den övergripande säkerhets- och effektprofilen för de tre studerade scheman.
Monoterapi
Data från två öppna, multicenter, randomiserade, kontrollerade kliniska prövningar med 1207 patienter stödjer användningen av XELODA i förstahandsbehandlingen av patienter med metastaserande kolorektalt karcinom. De två kliniska studierna var identiska i design och genomfördes i 120 centra i olika länder. Studie 1 genomfördes i USA, Kanada, Mexiko och Brasilien; Studie 2 genomfördes i Europa, Israel, Australien, Nya Zeeland och Taiwan. Totalt, i båda studierna randomiserades 603 patienter till behandling med XELODA 500 mg i en dos på 1250 mg/m2 två gånger dagligen under 2 veckor följt av en 1-veckors viloperiod och gavs som 3-veckorscykler; 604 patienter randomiserades till behandling med 5-FU och leukovorin (20 mg/m2 leukovorin IV följt av 425 mg/m2 IV bolus 5-FU, dag 1 till 5, var 28:e dag).
båda försöken utvärderades total överlevnad, tid till progression och svarsfrekvens (fullständig plus partiell respons). Svaren definierades av Världshälsoorganisationens kriterier och skickades till en blindad oberoende granskningskommitté (IRC). Skillnader i bedömningar mellan utredaren och IRC förenades av sponsorn, blindad för behandlingsarmen, enligt en specificerad algoritm. Överlevnad bedömdes baserat på en icke-underlägsenhetsanalys.
Baslinjedemografin för XELODA- och 5-FU/LV-patienter visas i Tabell 13.
Effektmålen för de två fas 3-prövningarna visas i och
Figur 3 Kaplan-Meier-kurva för total överlevnad av poolade data (studier 1 och 2)
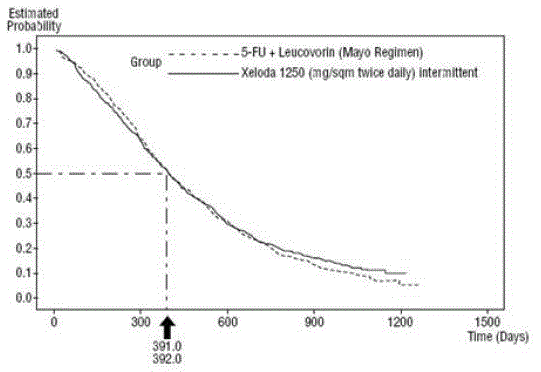
XELODA var överlägset 5-FU/LV för objektiv svarsfrekvens i studie 1 och studie 2. Likheten mellan XELODA 500 mg och 5-FU/LV i dessa studier utvärderades genom att undersöka den potentiella skillnaden mellan de två behandlingarna. För att säkerställa att XELODA 500mg har en kliniskt meningsfull överlevnadseffekt, utfördes statistiska analyser för att bestämma procentandelen av överlevnadseffekten av 5-FU/LV som bibehölls av XELODA. Uppskattningen av överlevnadseffekten av 5-FU/LV härleddes från en metaanalys av tio randomiserade studier från den publicerade litteraturen som jämförde 5-FU med regimer av 5-FU/LV som liknade kontrollarmarna som användes i dessa studier 1 och 2. Metoden för att jämföra behandlingarna var att undersöka det värsta fallet (95 % konfidens övre gräns) för skillnaden mellan 5-FU/LV och XELODA 500 mg, och att visa att förlust på mer än 50 % av 5- FU/LV-överlevnadseffekt uteslöts. Det visades att procentandelen av överlevnadseffekten av 5-FU/LV som bibehölls var minst 61 % för studie 2 och 10 % för studie 1. Det sammanslagna resultatet överensstämmer med en retention på minst 50 % av effekten av 5 -FU/LV. Det bör noteras att dessa värden för bevarad effekt är baserade på den övre gränsen för skillnaden mellan 5-FU/LV och XELODA 500 mg. Dessa resultat utesluter inte möjligheten av sann likvärdighet mellan XELODA 500 mg och 5-FU/LV (se och Figur 3 ).
Bröstcancer
XELODA har utvärderats i kliniska prövningar i kombination med docetaxel (Taxotere®) och som monoterapi.
I kombination med Docetaxel
Dosen av XELODA som användes i den kliniska fas 3-prövningen i kombination med docetaxel baserades på resultaten från en fas 1-studie, där en rad doser av docetaxel administrerade i 3-veckorscykler i kombination med en intermittent regim av XELODA (14 dagar) behandling, följt av en 7-dagars viloperiod) utvärderades. Kombinationsdosregimen valdes baserat på tolerabilitetsprofilen för de 75 mg/m2 som administrerats i 3-veckorscykler av docetaxel i kombination med 1250 mg/m2 två gånger dagligen under 14 dagar av XELODA 500 mg administrerat i 3-veckorscykler. Den godkända dosen på 100 mg/m2 docetaxel administrerad i 3-veckorscykler var kontrollarmen i fas 3-studien.
XELODA i kombination med docetaxel utvärderades i en öppen, multicenter, randomiserad studie i 75 centra i Europa, Nordamerika, Sydamerika, Asien och Australien. Totalt 511 patienter med metastaserad bröstcancer som var resistenta mot, eller återkommande under eller efter en antracyklininnehållande terapi, eller med skov under eller återkommande inom 2 år efter avslutad antracyklininnehållande adjuvant terapi. Tvåhundrafemtiofem (255) patienter randomiserades till att få XELODA 1250 mg/m2 två gånger dagligen i 14 dagar följt av 1 vecka utan behandling och docetaxel 75 mg/m2 som en 1-timmes intravenös infusion administrerad i 3-veckorscykler. I monoterapiarmen fick 256 patienter docetaxel 100 mg/m2 som en 1-timmes intravenös infusion administrerad i 3-veckorscykler. Patientdemografi finns i Tabell 16.
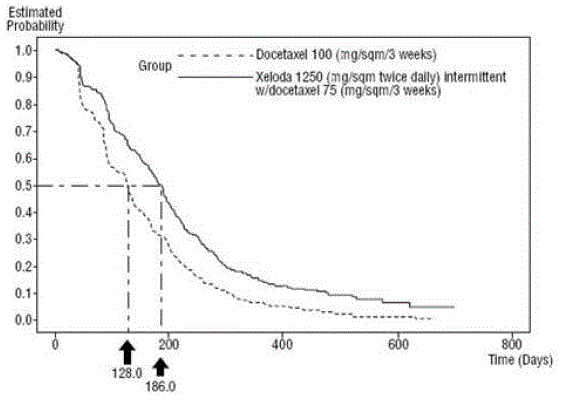
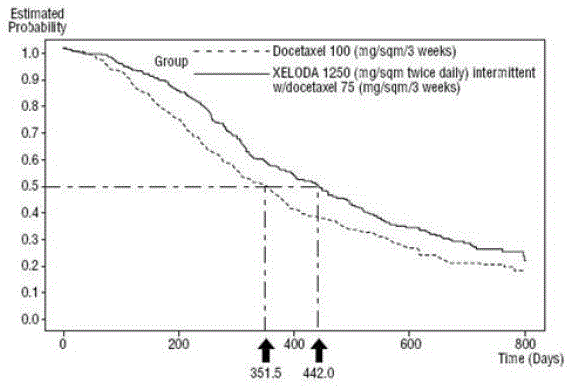
XELODA i kombination med docetaxel resulterade i statistiskt signifikant förbättring av tid till sjukdomsprogression, total överlevnad och objektiv svarsfrekvens jämfört med monoterapi med docetaxel som visas i och Bild 5 .
Figur 4 Kaplan-Meier uppskattningar för tid till sjukdomsprogression XELODA och Docetaxel vs Docetaxel
Figur 5 Kaplan-Meier uppskattningar av överlevnad XELODA och Docetaxel vs Docetaxel
Monoterapi
Antitumöraktiviteten av XELODA som monoterapi utvärderades i en öppen enarmad studie som genomfördes i 24 centra i USA och Kanada. Totalt 162 patienter med stadium IV bröstcancer inkluderades. Det primära effektmåttet var tumörsvarsfrekvens hos patienter med mätbar sjukdom, med svar definierat som en ≥50 % minskning av summan av produkterna av de vinkelräta diametrarna av bidimensionellt mätbar sjukdom under minst 1 månad. XELODA administrerades i en dos av 1255 mg/m2 två gånger dagligen under 2 veckor följt av en 1 veckas viloperiod och gavs som 3 veckors cykler. Baslinjedemografin och kliniska egenskaper för alla patienter (n=162) och de med mätbar sjukdom (n=135) visas i . Resistens definierades som progressiv sjukdom under behandling, med eller utan ett initialt svar, eller återfall inom 6 månader efter avslutad behandling med en antracyklin-innehållande adjuvant kemoterapiregim.
Antitumörsvar för patienter med sjukdomsresistenta mot både paklitaxel och ett antracyklin visas i
För subgruppen av 43 patienter som var dubbelt resistenta var mediantiden till progression 102 dagar och medianöverlevnaden 255 dagar. Den objektiva svarsfrekvensen i denna population stöddes av en svarsfrekvens på 18,5 % (1 CR, 24 PR) i den totala populationen av 135 patienter med mätbar sjukdom, som var mindre resistenta mot kemoterapi (se ). Mediantiden till progression var 90 dagar och medianöverlevnaden var 306 dagar.
PATIENTINFORMATION
XELODA® (zeh-LOE-duh) (capecitabin) tabletter
Vilken är den viktigaste informationen jag borde veta om XELODA 500mg?
XELODA 500mg kan orsaka allvarliga biverkningar, inklusive:
- XELODA kan interagera med blodförtunnande läkemedel, såsom warfarin (COUMADIN®). Att ta XELODA 500 mg tillsammans med dessa läkemedel kan orsaka förändringar i hur snabbt dina blodproppar och kan orsaka blödningar som kan leda till döden. Detta kan inträffa så snart som några dagar efter att du börjar ta XELODA 500 mg, eller senare under behandlingen, och möjligen till och med inom 1 månad efter att du slutat ta XELODA. Din risk kan vara högre eftersom du har cancer och om du är över 60 år.
- Innan du tar XELODA, berätta för din läkare om du tar warfarin (COUMADIN) eller annat blodförtunnande läkemedel.
- Om du tar warfarin (COUMADIN) eller annat blodförtunnande medel som liknar warfarin (COUMADIN) under behandling med XELODA, bör din läkare ta blodprov ofta för att kontrollera hur snabbt dina blodproppar under och efter att du avslutar behandlingen med XELODA. Din vårdgivare kan ändra din dos av det blodförtunnande läkemedlet om det behövs.
Ser "Vilka är de möjliga biverkningarna av XELODA 500mg?" för mer information om biverkningar.
Vad är XELODA?
XELODA är ett receptbelagt läkemedel som används för att behandla personer med:
- cancer i tjocktarmen som har spridit sig till lymfkörtlar i området nära tjocktarmen (Dukes C-stadium), efter att de opererats.
- cancer i tjocktarmen eller ändtarmen (kolorektal) som har spridit sig till andra delar av kroppen (metastaserande), som din första behandling av din cancer i detta skede.
- bröstcancer som har spridit sig till andra delar av kroppen (metastaserande) tillsammans med ett annat läkemedel som kallas docetaxel efter behandling med vissa andra läkemedel mot cancer har inte fungerat.
- bröstcancer som sprids till andra delar av kroppen och som inte har blivit bättre efter behandling med paklitaxel och vissa andra cancerläkemedel, eller som inte kan få någon mer behandling med vissa läkemedel mot cancer.
Det är inte känt om XELODA 500mg är säkert och effektivt för barn.
Ta inte XELODA om du:
- har allvarliga njurproblem
- är allergiska mot capecitabin, 5-fluorouracil eller något av dess ingredienser i XELODA. Se slutet av denna bipacksedel för en komplett lista över ingredienser i XELODA.
Prata med din vårdgivare innan du tar XELODA om du är osäker på om du har något av tillstånden som anges ovan.
Innan du tar XELODA, berätta för din vårdgivare om alla dina medicinska tillstånd, inklusive om du:
Ser "Vad är den viktigaste informationen jag borde veta om XELODA?"
- har haft hjärtproblem
- har njur- eller leverproblem
- har fått veta att du saknar enzymet DPD (dihydropyrimidindehydrogenas).
- är gravid eller planerar att bli gravid. XELODA 500 mg kan skada ditt ofödda barn. Din vårdgivare bör göra ett graviditetstest innan du påbörjar behandling med XELODA. Berätta genast för din vårdgivare om du blir gravid eller tror att du kan vara gravid under behandling med XELODA.
- Kvinnor som kan bli gravida bör använda effektiv preventivmedel under behandlingen och i 6 månader efter den sista dosen. Prata med din vårdgivare om preventivmedel som kan vara rätt för dig under behandling med XELODA.
- Män som har kvinnliga partners som kan bli gravida bör använda effektiv preventivmedel under behandlingen och i 3 månader efter den sista dosen.
- ammar eller planerar att amma. Det är inte känt om XELODA går över i din bröstmjölk. Amma inte under behandling med XELODA 500 mg och under 2 veckor efter den sista dosen.
Berätta för din vårdgivare om alla läkemedel du tar, inklusive receptbelagda och receptfria läkemedel, vitaminer och växtbaserade kosttillskott. XELODA kan påverka hur andra läkemedel fungerar och andra läkemedel kan påverka hur XELODA verkar.
Vet vilka mediciner du tar. Håll en lista över dem för att visa din vårdgivare och apotekspersonal när du får ett nytt läkemedel.
Hur ska jag ta XELODA 500mg?
- Ta XELODA exakt som din läkare säger åt dig att ta det.
- Din vårdgivare kommer att tala om för dig hur mycket XELODA du ska ta och när du ska ta den.
- Ta XELODA 2 gånger om dagen, 1 gång på morgonen och 1 gång på kvällen.
- Ta XELODA 500 mg inom 30 minuter efter avslutad måltid.
- Svälj XELODA 500 mg tabletter hela med vatten. Låt bli krossa eller skär XELODA 500 mg tabletter. Om du inte kan svälja XELODA tabletter hela, berätta för din sjukvård.
- Din vårdgivare kan ändra din dos, tillfälligt avbryta eller permanent avbryta behandlingen med XELODA 500 mg om du utvecklar biverkningar.
- Om du tar för mycket XELODA, ring din vårdgivare eller gå till närmaste akutmottagning direkt.
Vilka är de möjliga biverkningarna av XELODA?
XELODA kan orsaka allvarliga biverkningar inklusive:
Illamående och kräkningar är vanliga med XELODA. Om du tappar aptiten, känner dig svag och har illamående, kräkningar eller diarré kan du snabbt bli uttorkad.
Sluta ta XELODA och ring din vårdgivare omedelbart om du:
Om ditt antal vita blodkroppar är mycket lågt löper du ökad risk för infektion. Ring din vårdgivare omedelbart om du får feber på 100,5 oF eller mer eller har andra tecken och symtom på infektion.
- Ser "Vad är den viktigaste informationen jag borde veta om XELODA?"
- Diarre. Diarré är vanligt med XELODA och kan ibland vara allvarligt. Sluta ta XELODA och ring din vårdgivare omedelbart om antalet tarmrörelser du har på en dag ökar med 4 eller fler tarmrörelser än vad som är vanligt för dig eller tarmrörelser på natten. Fråga din vårdgivare om vilka läkemedel du kan ta för att behandla din diarré. Om du har svår blodig diarré med svår buksmärta och feber, sluta ta Xeloda 500 mg och ring din vårdgivare eller gå till närmaste akutmottagning direkt.
- Hjärtproblem. XELODA kan orsaka hjärtproblem, inklusive: hjärtinfarkt och minskat blodflöde till hjärtat, bröstsmärtor, oregelbundna hjärtslag, förändringar i ditt hjärtas elektriska aktivitet som ses på ett elektrokardiogram (EKG), problem med din hjärtmuskel, hjärtsvikt och plötslig död. Sluta ta XELODA 500 mg och ring din vårdgivare eller gå till närmaste akutmottagning omedelbart om du får några nya symtom på ett hjärtproblem, inklusive:
- bröstsmärta
- yrsel
- andnöd
- yrsel
- Förlust av för mycket kroppsvätska (uttorkning) och njursvikt. Uttorkning kan inträffa med XELODA 500 mg och kan orsaka plötslig njursvikt som kan leda till döden. Du löper högre risk om du har njurproblem innan du tar XELODA 500 mg och även tar andra läkemedel som kan orsaka njurproblem.
- kräks 2 eller fler gånger på en dag.
- bara kan äta eller dricka lite då och då, eller inte alls på grund av illamående.
- har diarré. Se "diarré" ovan.
- Allvarliga hud- och munreaktioner.
- XELODA 500mg kan orsaka allvarliga hudreaktioner som kan leda till döden. Berätta omedelbart för din vårdgivare om du får hudutslag, blåsor och flagnande hud. Din vårdgivare kan säga åt dig att sluta ta XELODA om du får en allvarlig hudreaktion. Ta inte XELODA 500 mg igen om detta händer.
- XELODA 500 mg kan också orsaka "hand- och fotsyndrom". Hand- och fotsyndrom är vanligt med XELODA 500 mg och kan orsaka domningar och förändringar i känsel i händer och fötter, eller orsaka rodnad, smärta, svullnad av händer och fötter. Sluta ta XELODA och ring din vårdgivare omedelbart om du har något av dessa symtom och du inte kan utföra dina vanliga aktiviteter.
- Hand- och fotsyndrom kan leda till förlust av fingeravtryck vilket kan påverka din identifiering.
- du kan få sår i munnen eller på tungan när du tar XELODA. Sluta ta XELODA 500 mg och ring din vårdgivare om du får smärtsam rodnad, svullnad eller sår i munnen eller tungan, eller om du har problem med att äta.
- Ökad nivå av bilirubin i ditt blod och leverproblem. Förhöjt bilirubin i ditt blod är vanligt med XELODA 500 mg och kan ibland också vara allvarligt. Din vårdgivare kommer att kontrollera dig för dessa problem under behandling med XELODA.
- Minskat antal vita blodkroppar, blodplättar och röda blodkroppar. Din vårdgivare kommer att ta blodprov under behandlingen med XELODA för att kontrollera dina blodkroppar.
Personer som är 80 år eller äldre kan vara mer benägna att utveckla allvarliga eller allvarliga biverkningar med XELODA.
De vanligaste biverkningarna av XELODA 500mg inkluderar:
- diarre
- smärta i magområdet (buk).
- hand- och fotsyndrom
- svaghet och trötthet
- illamående
- kräktes
- ökade mängder nedbrytningsprodukter för röda blodkroppar (bilirubin i ditt blod).
Allvarliga allergiska reaktioner kan inträffa med XELODA. Berätta för din vårdgivare om du någonsin har haft en allergisk reaktion mot capecitabin eller 5-fluorouracil. Ser ”Ta inte XELODA om du:”. Sluta ta XELODA 500 mg och ring din vårdgivare omedelbart eller gå till en akutmottagning om du har något av följande symtom på en allvarlig allergisk reaktion:
- röda kliande nässelutslag på huden (nässelutslag)
- hudrodnad
- svullnad av ansikte, läppar, tunga eller svalg
- utslag
- klåda
- problem med att svälja eller andas
XELODA 500 mg kan orsaka fertilitetsproblem hos kvinnor och män. Detta kan påverka förmågan att få barn. Prata med din vårdgivare om du är orolig för fertilitet.
Dessa är inte alla möjliga biverkningar av XELODA.
Ring din läkare för medicinsk rådgivning om biverkningar. Du kan rapportera biverkningar till FDA på 1-800-FDA-1088.
Hur ska jag förvara XELODA?
- Förvara XELODA 500 mg i rumstemperatur mellan 68°F till 77°F (20°C till 25°C).
- Förvara XELODA 500mg i en väl tillsluten behållare.
- Fråga din vårdgivare eller apotekspersonal hur du säkert kastar oanvänd XELODA.
Förvara XELODA och alla läkemedel utom räckhåll för barn.
Allmän information om säker och effektiv användning av XELODA.
Läkemedel skrivs ibland ut för andra ändamål än de som anges i en patientinformationsbroschyr. Använd inte XELODA för ett tillstånd som det inte har ordinerats för. Ge inte XELODA 500 mg till andra personer, även om de har samma symtom som du har. Det kan skada dem. Du kan fråga din apotekspersonal eller vårdgivare om information om XELODA 500mg som är skriven för vårdpersonal.
Vilka är ingredienserna i XELODA 500mg?
Aktiv beståndsdel: capecitabin
Inaktiva Ingredienser: vattenfri laktos, kroskarmellosnatrium, hydroxipropylmetylcellulosa, mikrokristallin cellulosa, magnesiumstearat och renat vatten. Beläggningen av persika eller ljus persika innehåller hydroxipropylmetylcellulosa, talk, titandioxid och syntetiska gula och röda järnoxider.
För mer information, gå till http://www.gene.com/patients/medicines/xeloda 500mg eller ring 1-877-436-3683.
Denna patientinformation har godkänts av US Food and Drug Administration.